Closing in on a cure

Colin Rensch fell in love with the piano when he was 11 years old. Seven years later, he got the chance to take a piano lesson from jazz legend Herbie Hancock, thanks to the Make-A-Wish Foundation.
Rensch was a percussionist in concert band at his high school in Mattawan, Michigan and then studied music history at Hope College in Holland, Michigan. Now 25 years old, he is finishing a master’s degree in history at Western Michigan University and applying to Ph.D. programs for musicology.
He also has Duchenne muscular dystrophy, the most common form of childhood-onset muscular dystrophy. When he was in high school, he began to lose strength in his arms and channeled his passion for music first into composing and then into studying the history of music.

“Knowing what I can and cannot do, that doesn’t really faze me very much. But it’s been a learning curve,” said Rensch, who has a number of assistants, sometimes up to 10, who help him throughout the day. “I first started with my assistants when I was in college, (and) I’ve learned a lot since then. I would say that coordinating with other people helping me is the most important thing.”
Duchenne, or DMD, is one of nine forms of muscular dystrophy, inherited diseases in which muscle tissue wastes away. DMD almost exclusively affects babies assigned male at birth, at an incidence of 1 in 3,500. Among muscular dystrophies, it is the most severe. Despite advances in cardiac and respiratory care that have allowed people with DMD to survive into adulthood, nearly all succumb to heart failure by their mid-30s.
DMD was first observed in the mid-19th century but was untreatable until corticosteroids were developed in the 1950s. For decades, steroids were the primary treatment for patients with DMD, along with drugs for secondary conditions that affect patients’ thyroid glands and lungs. That changed in 2016 with the rollout of Sarepta Therapeutics’ Exondys 51, the brand name for eteplirsen, a drug that restores limited expression of the dystrophin protein by patching, or skipping, over the coding region of DNA called exon 51, which is missing in 13% of Duchenne patients. The drug, approved by the Food and Drug Administration in 2016, is estimated to cost $300,000 per year.

But exon skipping is just one path forward for treating the disease. Each of the pharmaceutical companies Solid Biosciences, Pfizer and Sarepta has a drug candidate in clinical trials that delivers microdystrophin, a pared-down version of the massive gene that codes for dystrophin, to the muscles of people with DMD, where it is translated to make a mostly functional version of the dystrophin protein.
At the same time, studies in a handful of labs have shown promise in using the gene-editing machinery CRISPR–Cas9 to correct DMD-causing mutations in increasingly complex animal models. If researchers can find ways to subvert the human immune system’s response to the viral vectors that deliver CRISPR–Cas9 and can produce enough of those vectors to meet the needs of patients with DMD and other monogenetic disorders, they may be able to restore functioning fibers to the muscles and hearts of more than 300,000 people worldwide.
Faulty fibers
Situated beneath the cell membranes of striated muscle fiber cells, dystrophin proteins support muscle strength. In their absence, the muscle fibers’ actin and myosin contract and expand normally but the fibers are damaged more easily by routine activity.
Before children with DMD begin walking, their muscles generally don’t undergo enough strain to show wear. This means diagnoses are usually made when a child is 2 to 3 years old. Rensch said he was diagnosed when he was about 3.
During early childhood, however, the muscular stem cells that repair muscle by creating new tissue are extremely active and can compensate for and mask the damage being done, delaying a diagnosis until a child is age 7 or older.
Jeff Chamberlain is a neurologist at the University of Washington with expertise in muscular dystrophies.
“Early on in life, the boys do pretty well because there’s all this ongoing muscle cell damage, but then it’s repaired,” Chamberlain said. “The problems start occurring around age 4 or 5. Patients start losing muscle mass, and what’s thought to be happening is that this repair process just cannot keep up any longer.”
As the repair process fails, macrophages that produce cytokines infiltrate inflamed muscle cells. This causes both the formation of excess fibrous tissue and the emergence of fat cells within the muscle tissue.

“There’s a sort of competition between fibrotic cells and fat cells versus the actual muscle stem cells, and, so, eventually the muscle regeneration starts losing out with these other processes,” Chamberlain said. “And the net result is over time patients lose muscle mass, and it’s replaced by connective tissue and fat cells.”
The loss of muscle mass is progressive. “I probably was ambulatory or … semiambulatory until I was about 13,” Rensch said. At that time, he started using a mobility scooter full time. He supplemented this with an elevating wheelchair when he started high school.
As the disorder progresses, patients also lose cardiomyocytes, a special subset of muscle cells that make up the heart.
DMD almost exclusively affects people with one copy of an X chromosome. The dystrophin gene is on the X chromosome — if someone has two copies, the body generally can compensate for the faulty gene with a functional one. Given the location of the causative gene, DMD most often is inherited from mothers who have one affected chromosome.
According to Eric Olson, a molecular biologist at the University of Texas Southwestern Medical Center who specializes in heart and muscle development, the unusually large size of the Duchenne gene makes routine prenatal screening largely impractical.
“It’s not easy,” Olson said. “If one does not know if there’s a mutation or where it might reside in this massive gene, it’s not trivial to screen for it.”
About one-third of cases develop spontaneously during gestation and are therefore unlikely to be caught by prenatal screening: If neither parent is a known carrier, they wouldn’t expect the disorder to develop during pregnancy.
It is, however, possible to screen for the disorder in newborns by measuring circulating levels of creatine kinase, a byproduct of muscular inflammation. “If it’s elevated tenfold, that’s diagnostic or highly indicative of Duchenne,” said Olson.

To help identify the disorder as early as possible, the Muscular Dystrophy Association has urged the Department of Health and Human Services to add a screen for DMD to the Recommended Uniform Screening Panel, a list that most states adopt for their universal newborn screening programs.
Skipping gaps
The Duchenne gene is made up of 79 exons. These are spliced together when noncoding introns are removed from pre-mRNA to make mature mRNA, which is translated to create the amino acids that make up the dystrophin protein. Which of the exons or interspersed introns are missing varies from patient to patient.
Some exons, such as exon 74, can be deleted without affecting the reading frame of adjacent exons. In the absence of exon 74, translation of exons 73 and 75 proceed uninterrupted, giving rise to a shorter but mostly functional dystrophin protein.
This is the case with Becker muscular dystrophy, a less severe disorder related to DMD that occurs in about one in 18,000 births. The body of a person with BMD produces dystrophin proteins with reduced functionality, and they may retain enough muscle mass to continue walking into their 40s and 50s.
In DMD, however, deletion of an exon disrupts the reading frame of mature mRNA, prematurely terminating translation and creating a completely nonfunctional dystrophin protein. This is what happens with exon 51, which Exondys 51 encourages transcriptional machinery to skip during pre-mRNA spicing by using DNA fragments known as antisense oligonucleotides. The result is a restored reading frame and a mostly functional dystrophin protein.
Whether a mutation on the dystrophin gene will give rise to DMD or BMD depends on which exons are affected by the mutation. However, sometimes mutations can present somewhere in the middle — as happened with Benjamin Dupree.

Dupree was 9 years old when doctors finally were able to explain that he had a hard time keeping up with other kids on the playground because he had a mild form of DMD; within six years, the condition progressed so that he was no longer able to walk on his own.
Rather than missing an exon, Dupree’s dystrophin gene codes for the insertion of an extra intron. That intron, 47, codes for a stop codon, which ends translation of the dystrophin protein prematurely. According to Dupree, a small portion of his dystrophin proteins remain functional, meaning his symptoms present in a middle ground between DMD and BMD.
Normally, “it becomes hard to lift your arms around age 20 or so,” said Dupree who is now 26. “That’s been a little bit less severe for me. I still am able to put my arms above my head and lift a decent amount of weight.”
Dupree has a bachelor’s degree in biochemistry from Southern Methodist University, and volunteers for Texas Scottish Rite Hospital for Children. He is preparing for a graduate program in social work, after which he hopes to help guide other young people with DMD through the challenges he’s faced. Since 2012, he has been a patient advocate for the advocacy group Parent Project Muscular Dystrophy, for which he first traveled to Washington, D.C., in 2014. He also has served on the FDA’s advisory committee for Exondys 51, though it is not a treatment option for his form of the disease.
“I try to look at it from the position of ‘If this drug was something that was applicable to every exon, in every case, how would I feel about that? What would my perspective be if this drug would work for me?’” he said.
Clinical trial data suggest that Exondys 51 works as intended, and at least half of the mutations that give rise to DMD could be amenable to exon skipping. Sarepta and other pharmaceutical companies have additional exon-skipping drugs in preclinical and clinical development for many of the missing exons that a faulty DMD gene may lack.
“I think that if there are enough (FDA approvals) for different exons or different drugs, it would build evidence for the approval of a mechanism rather than just individual drugs,” Dupree said. “I think that’s where they could eventually lead to something that would help me.”
However, despite the FDA’s approval of Exondys 51 in 2016 and recent clinical evidence that Exondys 51 slows patients’ respiratory decline, a recent report published by the Institute for Clinical and Economic Review claimed that Sarepta had not collected sufficient evidence that the drug benefits patients.
These concerns played out in August when the FDA denied approval of Sarepta’s second exon-skipping drug, Vyondys 53, designed for the exon 53 deletion that causes 8% of DMD cases.
All exon-skipping drugs must be administered continuously, which compounds their high price tag.
But re-administration may not be necessary if adeno-associated viral vectors, or AAVs, are used to smuggle smaller versions of an otherwise massive gene into patients’ muscles.
Microdystrophin delivery
At 2.3 megabases, the dystrophin gene is the largest in the human genome; the median size of a human gene is 26 kilobases. The most commonly used vectors for gene therapies, AAVs have a capacity for carrying genetic material just shy of 5 kB, which can be upped to 9 kB by using dual-vector systems.
Overall size isn’t a direct proxy for the size of a resultant protein or even the size of the mature mRNA generated once introns have been excised. For example, the gene that codes for the largest human protein, the muscle spring protein titin, contains 365 exons but is only around 0.3 mB long. In the case of dystophin, massive introns make up more than 99% of the gene.
The relatively small size of the dystrophin gene’s exons made it possible for Jeff Chamberlain and his colleagues at the University of Washington to develop microdystrophin, a compact version of the dystrophin gene around 4 kb long that codes for a smaller yet still functional version of the dystrophin protein. A similar, slightly larger iteration called minidystrophin subsequently was developed by a separate research group and is being evaluated in clinical trials conducted by Pfizer.
The AAV-based system delivers a copy of the microdystrophin gene flanked with promoter and enhancer sequences, which, respectively, help initiate transcription of a stretch of DNA and increase its odds of being transcribed. The promoter and enhancer sequences, which are specific to muscle cells, were developed by Chamberlain and his colleague Steve Hauschka.
“If you delivered the same proteins with a ubiquitous promoter that was active in many cells in the body, you could often get a very potent and sometimes lethal immune response,” Chamberlain said. “So we’ve focused a little more on trying to restrict gene expression to muscle.”

The muscle-specific promoters turned out to be a boon for drug development. The microdystrophin drugs developed by Pfizer, Sarepta and Solid Bioscience and now in early clinical trials all use muscle-specific enhancers developed by Chamberlain and Hauschka.
Dupree, whose symptoms fall between BMD and DMD and whose muscles contain truncated dystrophin proteins due to an intron insertion, said he is optimistic about the prospect of drugs that can induce the production of microdystrophin. Unlike exon-skipping therapies, a single microdystrophin drug would work for people with different DMD-causing mutations.
“I think the microdystrophin is a really great option for us,” he said. “I don’t think that it’s going to ever really be a complete solution. But I think it will be an improvement on what’s currently here that’ll hold people over until we have a more permanent or effective solution.”
Despite the increase of expression in skeletal muscle, though, microdystrophin therapies still run into an obstacle many drugs face in the body — the liver.
According to Luk Vandenberghe, a professor at Harvard’s Grousbeck Gene Therapy Center specializing in AAV production, most AAVs that enter the body ultimately wind up in the liver rather than in the organ or tissue for which their genetic payload is intended.
“We believe that more than 90% of that dose ultimately gets trapped by the liver, which is a tissue that we don’t need to target,” Vandenberghe said.
Small virus, giant reactors
This problem of viral misdirection extends to treatments that might use CRISPR–Cas9, as that system also is delivered in AAVs.

CRISPR, which stands for clustered regularly interspersed short palindromic repeats, first evolved as a bacterial defense mechanism that allows microorganisms to fight off hostile viruses by storing their genetic information alongside a set of enzymes known as CRISPR-associated proteins, or Cas, that are highly effective at snipping DNA. By turning the stored viral DNA into guide RNAs that complex with Cas, the bacteria cells can target and destroy a matching virus’ DNA before it can replicate.
Currently, there are no methods for removing genome-editing tools from the body once they’ve been introduced, a concern of Vandenberghe’s.
“One needs to have a short-term intervention … and then these genome-editing tools ideally need to leave the scene, because if they linger around for too long, they could have their own set of problems,” such as off-target effects, Vandenberghe said.
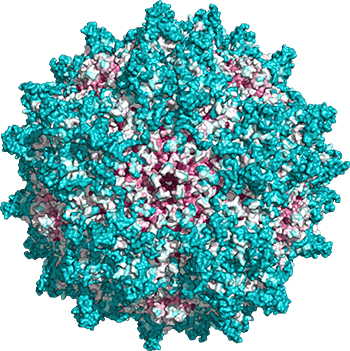
Even if early clinical trials don’t raise safety concerns, difficulty with large-scale production of AAVs remains a major obstacle.
“It’s just hard to make,” Vandenberghe said. “People are looking at generating 100,000-, 200,000-liter bioreactors, as large as you can think to get to the amount of virus needed.”
By his estimates and those of his colleagues, the amount of AAVs needed to deliver a single dose of CRISPR–Cas9 into someone with DMD would cost hundreds of thousands of dollars.
To produce the many subtypes of AAVs used in basic and preclinical research involving CRISPR, manufacturers reprogram cells, such as the mammalian cell line HEK293 or the insect cell line Sf9, to act as small viral factories. Whatever the cell line, each cell can produce about 100,000 viral particles. By Vandenberghe’s account, a single dose of AAVs is about 100 trillion viral particles for each kilogram the patient weighs. After factoring in weight, that comes to at least 10 orders of magnitude more than what a single cell can produce.
“If you have to treat the global population of DMD patients or even just the population in the Western world, we are talking in scales that the industry hasn’t seen,” he said. “So we need innovation there. And that innovation is only slowly coming forward.”

“There are efforts under way to have different (AAV) serotypes, so that maybe you’ve been exposed to one and you could come back a second time with an alternative serotype that would not have a preexisting immunity,” he said. “There may be ways to combine that with immunosuppression or other approaches.”
Mending fibers
Over the past five years, Olson’s lab in Dallas has used CRISPR–Cas9 to repair the mutations underlying DMD in mice and in canines, which are a better proxy for the human form of the disease than mice.

At the same time, Charles A. Gersbach and colleagues at Duke University have been using a CRISPR–Cas9 system to edit the dystrophin gene in mouse models. In a paper published in 2016 in the journal Science, the researchers repaired a gene with induced DMD mutations, turning it to a variant that resembled BMD. Gersbach’s paper appeared as one of a trio of articles in Science by teams at Duke, at Harvard University (led by the cell biologist Amy Wagers), and at UT Southwestern Medical Center (led by Olson) that described treating DMD in mice with CRISPR–Cas9.
Olson’s papers on correcting DMD mutations in mice caught the eye of Debra Miller, CEO of the nonprofit CureDuchenne. Together, Miller and Olson founded Exonics Therapeutics, a biotechnology company with the express goal of using CRISPR–Cas9 technology licensed from Olson’s lab to develop a platform for correcting DMD in humans.

Olson and his lab now are working out how to correct as many of the 3,000 mutations that can cause DMD as possible with a single CRISPR–Cas9 system.
“That’s a big challenge when it comes to CRISPR — how can you correct all these different mutations by gene editing?” Olson said.
Many mutations that cause DMD cluster in specific regions of the genome and can be targeted en masse. One method that takes advantage of this phenomenon, single-cut CRISPR, was developed by Olson and his colleagues.
“We can make a single cut in the DNA in regions around these hot spots, and that allows for either skipping of an exon that’s out of frame and restoring the reading frame or … we can reframe the protein by insertion of a single nucleotide at the cut site,” Olson said.
That process is conceptually similar to exon skipping: In the 6% of DMD patients missing exon 44, exons 43 and 45 become spliced together during translation to mRNA, which creates a premature termination codon that disrupts the reading frame of that mRNA, ending translation of the protein.
By using CRISPR–Cas9 to insert a single missing nucleotide, or delete two, the triplet codons that code for amino acids, like the stop codon, can be reframed properly, restoring translation and function of the protein.
However, the composition of skeletal muscles complicates the repair of dystrophin proteins in muscle cells. While typical cells consist of a nucleus and organelles suspended in cytoplasm and wrapped in a cell wall, muscle cells are made of striated bands of proteins with several nuclei. This allows them to perform their essential flexing and contracting but means they don’t divide. Instead, as muscle cells are damaged and die off, they are replaced by new cells, meaning any changes to them won’t be permanent.
Enter stem cells.
When muscle cells die, the myogenic stem cells, or satellite cells, that sit beneath the lowest layer of skin are activated to create new muscle cells. By targeting this source of muscle fibers, Amy Wagers and colleagues at the Harvard Stem Cell Institute believe that genome-level repairs to the dystrophin gene could be made permanent and produce the full-length proteins that microdystrophin approximates.
“If you don’t also modify the gene in satellite cells, in the sense that these cells seed replacement of the muscle fibers, there’s the possibility that, over time, the modified genes that are therapeutic will get diluted in muscle by the fusion into the fibers of new, unmodified satellite cells,” Wagers said.
In a paper published in the journal Cell Reports in June, Wagers and colleagues reported that when satellite cells in mice were targeted with a variety of muscle-specific adeno-associated viruses containing a gene that codes for the Cre protein, which is able to activate a simple fluorescent gene-editing reporter system by cutting at Lox sites, the fluorescent protein was expressed in more than 60% of the cells.
“We looked to see whether we were also getting modification in the precursors to muscle fibers in the muscle stem cells,” Wagers said. “And we got evidence for that using two different genetic systems, that one could modify the DMD gene in satellite cells.”
In addition to the need to validate the safety and efficacy of the CRISPR–Cas9 system, Wagers has concerns about the immune system’s tendency to clear adeno-associated viruses from the body after its first encounter with them.
“Can you get enough efficiency with a single dose? Or can you somehow circumvent the immune response so that you can do multiple dosing?” she said. “Those are sort of related challenges.”
Repair versus replace
The treatment that works for each patient ultimately may come down to their mutation.
“Every patient has a different mutation,” Chamberlain said. “And, importantly, 70% of all patients have deletion mutations or duplication mutations … they’re missing large portions of the genes, which means they’re missing large portions of the coding region. So, if you apply gene editing to those patients, you’re going to make a minidystrophin or microdystrophin.”
For point mutations, in which the reading frame that transcribed mRNA is shifted out of place, a repair might create a highly functional dystrophin that works more effectively than a microdystrophin, Chamberlain said. “But in other cases where there’s a larger deletion or duplication, or the deletion removes a critical functional domain of dystrophin, then it’s very likely that the microdystrophin would work better than that repaired copy.”
According to Wagers, repairing and replacement treatments might end up being complementary.
“I have seen people sort of spin this as a little bit as gene therapy versus gene editing … and that’s, I think, a false dichotomy,” Wagers said. “The combination could be even more powerful because you have the immediate expression of the microdystrophin, which might stabilize fibers in which editing is going on, in order to ensure a higher rate of editing.”
When muscle fibers are dystrophic, only a fraction of the nuclei inside of them can be edited. This carries the possibility that fibers die even though nuclei have been edited, because the gene product — a functional dystrophin — hasn’t been expressed to rescue them yet.
“I think there’s opportunity for combination type of approaches with gene complementation … as a strategy for something that will give you both a transient and a long-term benefit,” she said.
Multiple deletions, multiple paths
Colin Rensch, the musicology student from Michigan, is among the 50% of DMD patients with multiple deletions; he is missing exons 46 through 52.
Since 2014, he, like Dupree, has been a patient advocate for Parent Project Muscular Dystrophy and has participated in federal advocacy efforts on Capitol Hill.
“I just think if you’re trying to get the most people affected by it, the microdystrophin seems (of) much better use” than exon skipping, Rensch said. “It would be great if one of these other treatments could help until CRISPR could be an option.”
As he makes plans to earn a doctoral degree in musicology, Rensch faces the possibility of moving away from the support system he and his family have in the Kalamazoo area. While assistants help him throughout the day, he lives independently in a house he and his family redesigned to be as accessible as possible.
“That’s the one huge advantage that I’d have to give up to go away to a different school,” he said. But his studies fall outside the graduate work offered by Western Michigan, so he’ll likely have to relocate.
Rensch is resilient and determined to continue his academic career.
“I think that, for me, that’s really an important thing — finding something meaningful in your life,” he said. “I have the same kind of dreams that anyone else does … I’m just going to keep living my life.”
Different dystrophies
Duchenne muscular dystrophy primarily affects people with one X chromosome but occurs in people with two X chromosomes, most assigned at birth as girls, at an incidence of 1 in 50 million. When someone has two X chromosomes, only one is turned on at a time in a cell, and this activity can vary throughout the body. Depending on where the mutated genes are expressed, a person with one copy of a Duchenne allele, or a manifesting carrier, might experience mild symptoms of muscular dystrophy. These symptoms can be similar to Becker muscular dystrophy, which also primarily affects boys, and manifests when mutations to the dystrophin gene produce truncated, but functional, dystrophin proteins. However, most of the other seven major forms of muscular dystrophy listed below more equally affect men, women and nonbinary people.
| Muscular dystrophy | Symptoms | Incidence | Typical age at onset |
| Myotonic | Myotonia, prolonged spasms or stiffness, in muscles after use. | 8 in 100,000 | Late childhood to middle age |
| Facioscapulohumeral | Weakness in the muscles that move the face, upper arm bone and shoulder blade. Can also affect speaking, walking, chewing and swallowing. | 4 in 100,000 | Young adulthood |
| Limb-Girdle | Weakness in the hips that moves to shoulders, arms and legs, with eventual loss of mobility. | 2 in 100,000 | Childhood to middle age, depending on type |
| Congenital | General muscle weakness. | 1 in 100,000 | Birth |
| Distal | Weakness in lower legs and arms, including hands, that may spread and progress to atrophy. | < 1 in 100,000 | Middle to old age |
| Emery-Dreifuss | Weakness in muscles of shoulders, upper arms and calves, in addition to joint stiffness and heart | < 1 in 100,000 | Childhood |
| Oculopharyngeal | Weakening of throat muscles, which progresses to interfere with swallowing. Also weakens eyelid muscles. | < 1 in 100,000 | Middle to old age |

Sickled cells and CRISPR controversies

CRISPR–Cas9, which first was demonstrated as a potential gene-editing technique by Jennifer Doudna and Emmanuelle Charpentier in 2012, can be used to edit the genome in two distinct ways.
In somatic genome editing, researchers edit defective genes in an organism’s affected tissues or in excised cells that are then reinserted. In germline editing, researchers aim to edit genes in embryos to prevent mutations from manifesting. Changes made during germline editing, including any potential errors introduced in the editing process, can be passed down to each successive generation.
In early 2017, the National Academy of Sciences and the National Academy of Medicine rolled out guidelines for how genome editing should proceed in humans. The academies essentially said researchers should proceed with extreme caution when editing the germline but could venture forth with somatic editing for diseases with well-understood monogenic causes.

“For something like sickle cell disease or Duchenne’s muscular dystrophy, these are diseases where there’s a single gene that is known to be mutated in patients that have the disorder,” Doudna said. “And the genetics of that are well worked out. So we understand that by making a change to that causative gene, one could actually mitigate the disease.”
Last November, Chinese scientist He Jiankui reported using CRISPR–Cas9 to remove from embryos the receptors to which HIV binds. His announcement at the Second International Summit on Human Genome Editing caused an international uproar.
In response to He’s actions and a Russian scientist’s recent declaration of intent to use CRISPR on viable embryos, the NAS, the NAM and the U.K. Royal Society convened another summit on genome editing in August, with a new report and guidelines to come next summer.
Meanwhile, clinicians in the U.S. recently moved ahead with somatic editing for sickle cell disease.
Sickle cell disease is caused by a genetic defect in which blood cells carry insufficient oxygen and stick to the inside of blood vessels. The disease affects millions of people worldwide, almost all of African descent. It causes debilitating pain and reduces lifespans; many patients die in their 40s. There are few treatment options beyond bone marrow transplants.
In July, doctors at HCA Healthcare’s TriStar Centennial Medical Center in Nashville administered to a patient with sickle cell disease bone marrow cells that had been edited to correct the defect that gives rise to faulty red blood cells. The clinical trial, conducted by the Boston-area companies Vertex Pharmaceuticals and CRISPR Therapeutics, will enroll up to 45 patients between the ages of 18 and 35.
Melissa Creary, a professor at the University of Michigan specializing in the societal history and impact of sickle cell disease in Brazil and the U.S., is cautiously optimistic about the development.
“I think that, generally, CRISPR sounds like an exciting new technology. But our excitement about new technology should be tempered,” Creary said.
“Who can afford it? How will people get access to it? What happens afterwards? What kind of support networks are in place? What are the unintended consequences? And how will people be supported around those unintended consequences if you’re marginalized, much less if you’re not? All of those questions, I think, need to be put in place much ahead of full implementation, while we’re thinking about the feasibility of the tech.”
The role of CureDuchenne

He recently graduated from the University of Southern California with a degree in journalism and is a multiplatform producer
for the Washington Post.
Over the past 16 years, CureDuchenne has provided seed funding to small biotech companies, helping secure more than $1.3 billion from larger pharmaceutical companies and venture capital firms; nine of its funded projects, including antifibrotic agents, exon skippers and gene-correcting agents, have gone on to clinical trials. CureDuchenne provided early funding for the research that led to Exondys 51 as well as Pfizer’s drug candidate using a minidystrophin.
Based in Newport Beach, California, CureDuchenne was co-founded in 2003 by Debra and Paul Miller less than a year after the couple found out that their son, Hawken, now 22, had Duchenne muscular dystrophy.
“We looked around, and there were a couple organizations, but they tended to be more support-type organizations and not focused on funding the cure,” Debra Miller said. “We never knew (about) venture philanthropy until after we started investing. We did all the due diligence and found out that, yes, legally, we can do this as a nonprofit, as long as we’re investing in our mission.”
In late 2015, Miller and her board began to take note of news about DMD and CRISPR–Cas9, which led her to Eric Olson at the University of Texas Southwestern Medical Center at Dallas.
Early meetings went well, and Olson and CureDuchenne Ventures founded Exonics Therapeutics. The company launched with $2 million in seed financing in February 2017 and recently was bought by the biopharmaceutical giant Vertex.
The acquisition, Olson said, “is going to greatly accelerate and enable our advancement of this technology in a way that would have been more challenging within a small startup biotechnology company.”
Miller is also optimistic about the impact Vertex will have on the scientific team’s research.
“They’re a powerhouse,” Miller said. “They know how to get drugs developed and approved. And I think if anybody can do this, Vertex will be the one to do it.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Catching tau in the act
Using a new proximity-labeling approach, researchers reveal how tangles of the brain-associated protein tau may disrupt RNA biology long before neurons die.
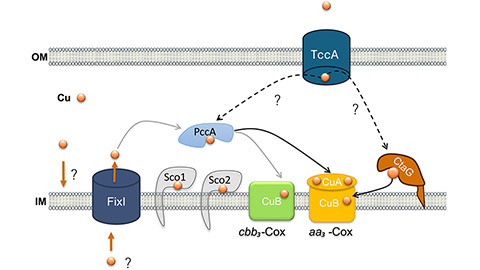
How copper delivery fuels bacterial respiration
Researchers identify the roles of several proteins in copper homeostasis in the aerobic bacterium Caulobacter vibrioides.
Revealing the glycoproteome of a cancer subtype
Researchers mapped the glycoproteome of extrahepatic cholangiocarcinoma and compared it to intrahepatic tumors. Differences in sugar modifications and immune cell content suggest new biomarkers and guide development of targeted immunotherapies.

Uncovering the mechanisms of a glycosylation disorder
Mutations in OGT, an enzyme that adds sugars to proteins, cause a rare neurological disorder. Using proteomics, researchers reveal how OGT interactions with TET proteins may trigger epigenetic changes and early neural defects.

Heat shock proteins as a promising breast cancer therapeutic
Researchers unveiled isoform-specific targets on heat shock protein 90 which may be beneficial in therapeutic development.

Optimized proteomic analysis of preserved biological tissue samples
Researchers have developed an optimized workflow for analyzing formalin-fixed paraffin-embedded tissue. This workflow provides an enhanced collection of unique proteins and phosphorylation sites for more detailed analysis of biological samples.