Defeating deletions and duplications

A small alteration on chromosome 15 can drastically affect development, influencing communication, motor skills, sleep and more. For families of children like Coral, who suffers from Dup15q syndrome, daily life involves navigating challenges such as seizures, communication barriers and the need for constant care.

Yet, amidst these challenges, researchers are making significant strides in understanding the molecular mechanisms behind 15q disorders and developing targeted therapies. Innovative approaches, including antisense oligonucleotide treatments, could one day be used to treat underlying genetic causes of conditions like Dup15q syndrome, offering the potential to transform patient care and improve quality of life for affected individuals and their families.
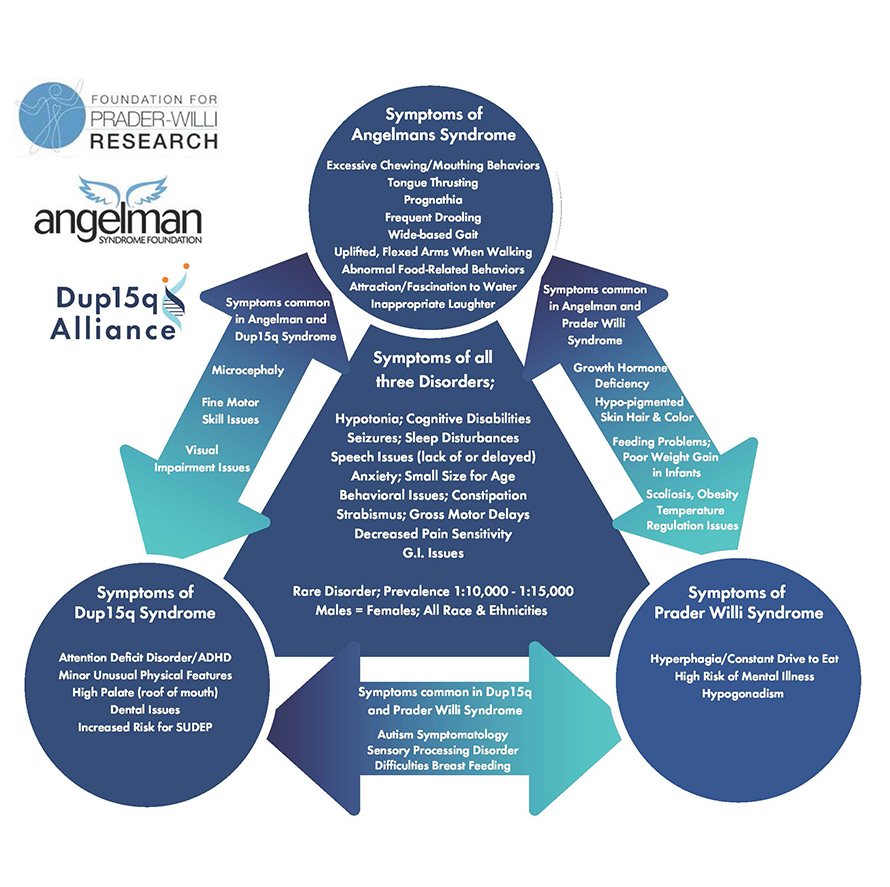
15q neurodevelopmental disorders comprise three distinct conditions: Angelman syndrome, Dup15q syndrome and Prader–Willi syndrome. Each is unique, yet all are caused by a change or mutation in the 15th chromosome. No cures have been identified for 15q disorders and treatment typically focuses on managing symptoms.
The National Library of Medicine estimates that Angelman syndrome affects one in every 20,000 people and Prader–Willi syndrome affects one in every 10,000 to 30,000. Dup15q has an unknown frequency, and it is estimated that it could be as high as one in every 5,000 people.
Prader–Willi Syndrome
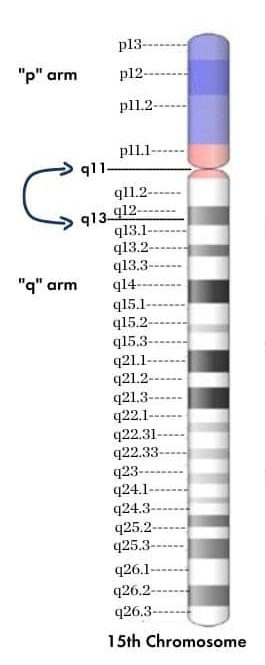
According to the Mayo Clinic, Prader–Willi syndrome, or PWS, is caused by a genetic mutation of undefined mechanisms. Current research and the National Institute of Child Health and Human Development suggest that this mutation causes an inability to express paternal genes. The critical region for PWS is on chromosome 15(q11–13). The disorder occurs when the paternal genes are not expressed due to a deletion, uniparental disomy (inheriting only maternal copies) or a defect in the imprinting center, a region that regulates parent-specific gene expression.
Typically, symptom presentation begins around two years of age, including feelings of persistent insatiable hunger, or hyperphagia, poor responsiveness, underdevelopment and decreased muscle tone, or hypotonia. Hyperphagia often leads to obesity, which means that individuals with the syndrome also maintain a higher risk of experiencing obesity-related complications such as Type II diabetes, high blood pressure, elevated cholesterol and heart disease. Additionally, decreased hormone production due to underdevelopment may lead to complications including sterility and osteoporosis.
Several therapeutic avenues have recently emerged, reporting promising results for PWS patients:
The drug, Vykat XR, produced by Soleno Therapeutics was recently approved by the Federal Drug Administration for PWS patients. It treats hyperphagia in patients aged four and older by activating potassium channels.
Harmony Biosciences recently received an Orphan Drug designation for Pitolisant, a therapeutic designed to treat excessive daytime sleepiness and behavioral disturbances. It works by increasing the histamine activity in the brain. This designation provides incentives for further testing, such as clinical trials testing the safety and efficacy of the drug in PWS patients.
Researchers at Duke University recently developed an epigenetic therapy using small molecule inhibitors to prevent H3K9 methylation in PWS patients, which enables expression of silenced maternal chromosomes to compensate for paternal deletion.
Angelman syndrome
Angelman syndrome, or AS, is often caused by abnormalities related to the ubiquitin ligase UBE3A gene on chromosome 15q. According to the Angelman Syndrome Foundation, this condition commonly occurs due to activation of only the maternal copy of the gene, with either a missing or defective paternal gene. However, AS can also be caused by the inheritance of two paternal genes. Symptoms of AS, including difficulty walking, lack of speech and seizures, typically first appear around six to 12 months of age.
A few avenues have recently emerged that may lead to AS treatments:
In 2024, Ionis Pharmaceuticals reported positive results for early clinical trials of a drug designed to unsilence the paternal UBE3A allele; thus, compensating for loss of function in the maternal copy of the gene in AS patients. After treatment, 65% of patients exhibited both cognitive improvements as well as enhanced fine motor skills.
The Foundation for Angelman Syndrome launched two biotech companies, MavriX Bio and CourageAS Bio, to advance therapeutic development for AS. MavriX focuses on developing the first AAV-delivered gene replacement therapy for AS, while CourageAS aims to develop a nonviral gene-editing therapeutic for patients.
Dup15q syndrome
The National Institutes of Health describes Dup15q syndrome, also referred to as maternal 15q duplication syndrome, as a condition caused by the presence of at least one extra chromosome 15 region (15q11.2-q13.1). Dup15q typically only occurs when the duplicate copy is maternally inherited.
Common characteristics of Dup15q syndrome include hypotonia, intellectual disabilities, autism spectrum disorder and epilepsy. Currently, treatment options for the condition are limited to the treatment of specific symptoms and surveillance, as well as genetic and prenatal testing to monitor development throughout pregnancy.
The 15q chromosome contains several regions known as segmental duplications, which have a higher susceptibility to rearrangement and thus mutation. Several genes of interest that may contribute to disease progression have been observed in this region, including the ubiquitin ligases UBE3A and HERC2 as well as neurotransmitter receptor components GABRB3, GABRA5 and GABRG3.
In 2023, Children’s Hospital Los Angeles launched Quindecim, a clinical trial focused on evaluating the safety and efficacy of Basmisanil, a drug that modulates neurotransmitter receptor activity, which can be overactive in Dup15q syndrome.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.