Before we’ve lost what we can’t rebuild: Hope for prion disease

“If you bring a loved one to a physician, will they give you prescriptions?” Eric Minikel asked. “Yes, but none of them are aimed at slowing (prion) disease.”

Eric Minikel and Sonia Vallabh, a husband-and-wife team, describe themselves as “on a lifelong quest to prevent prion disease.” They run a lab at the Broad Institute, studying prion disease, a rare, aggressive and fatal neurodegenerative disease that affects 300 people in the U.S. annually. All together, they do research, raise funds, collaborate with pharma companies, educate patients, physicians and genetic counselors and maintain a patient registry.
Their motivation comes from their own experiences. Vallabh carries a genetic predisposition to this disease and could develop symptoms at any time, and currently, there is no preventative, treatment or cure. To raise awareness, they founded the Prion Alliance.
Vallabh and Minikel collaborated with the biotech company Ionis Pharmaceuticals to develop a potential prion disease treatment and preventative. In December 2024, Ionis announced that an early clinical trial of ION717, the product of their collaboration, was fully enrolled.
When proteins turn against the brain
Prion disease is a family of diseases including Creutzfeldt–Jakob disease, sometimes referred to as “mad cow” disease, as well as Fatal Familial Insomnia and Gerstmann–Sträussler–Scheinker disease.
These diseases are united by the misfolding of prion protein, or PrP, in the brain. Normally folded, this neuronal cell surface glycoprotein plays a role in cell adhesion, signaling, stress protection and more. Once PrP misfolds, it induces surrounding PrP misfolding, triggering a cascade of misfolded protein that spreads and causes widespread cell death.
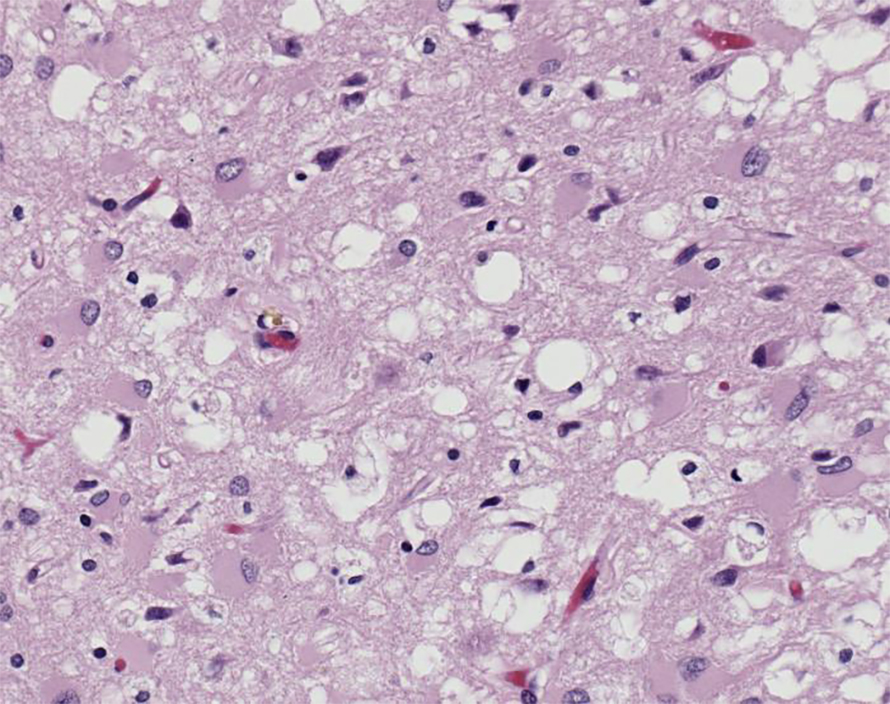
However, the exact mechanism of this harm is unknown. PrP may twist and damage the membrane’s structure as it misfolds or gets internalized by the cell, triggering the unfolded protein response and disrupting synaptic transmission. Misfolded PrP also aggregates, forming fibrils which may also induce neurotoxicity.
The term “prion protein” has two meanings. On one hand, it is a protein, PrP, encoded by the PRNP gene. A prion protein, however, can also be a term for any protein that, when misfolded, can induce the misfolding of other copies of the same protein, causing widespread misfolded proteins.
On average, prion diseases induce patient death within five months of the first symptom. Due to the rarity of the disease and symptom overlap with other neurodegenerative diseases, the diagnostic odyssey can be so long that a patient may be diagnosed with only weeks left to live or after autopsy.
Vallabh’s mother died in 2011 after a mysterious and rapid decline, which was identified as prion disease only during her autopsy.
“If you’ve seen someone you love go through this, you know how much is lost every single day.” Vallabh said. “To me, this is the biggest issue in neurodegeneration; we are losing things we have no idea how to rebuild. It’s particularly extreme in prion disease. It moves so fast.”
The charge
Watching her mother’s illness and the subsequent discovery that Vallabh carries a gene predisposing her to prion disease, upended Vallabh and Minikel's lives.
They left their careers in law and engineering, obtained Ph.D.s and reinvented themselves as prion disease researchers and advocates.
“We are coming at this with an unusual amount of specificity and focus,” Vallabh said. “We don’t have [a lot of] time. We have the mandate and the opportunity to be the people who lead the charge.”
Targeting the molecular cause
National Institutes of Health
About 85% of prion disease cases occur sporadically and are “totally random bad luck,” Minikel said. “For decades people searched for some link, maybe exposure, geography, family.” But it truly seems stochastic, developing in the brain for unknown reasons, he said.
The other 15% of cases are genetic, like Vallabh’s, and are caused by genetic alterations, such as missense mutations or insertions, in the PrP gene, PRNP, that make it more likely to misfold. The disease trigger is unknown; however, some scientists speculate that symptoms may be set off by oxidative stress or inflammation.
In addition, prion disease can be contracted from an external source such as diseased meat or contaminated surgical instruments. However, less than 1% of reported cases develop via this route.
While normal PrP may have a beneficial role in the brain, like helping maintain myelination, the disease seems to be caused by the accumulation of misfolded protein, not the loss of normal PrP. From a therapeutic perspective, Minikel said, this means lowering the levels of PrP protein in the brain could help patients.
“If you know what the disease is that you’re up against, the best thing you can do is go after the root molecular cause,” Minikel said. “People have tried to chase downstream things like neuroinflammation, but you’re chasing after a train that’s already left the station.”
Scientists have known for decades that animals lacking Prnp are fully protected from infectious prion disease. However, deleting a gene, which can be difficult in humans, isn’t the only way to eliminate the pathogenic protein.
Instead, Minikel and Vallabh’s ION717 aims to lower PrP levels by reducing its messenger RNA, or mRNA, and preventing protein translation.
Thwarting the prion protein
Vallabh and Minikel first connected with Ionis in 2014 while pursuing their Ph.D.s, beginning a collaboration to develop a treatment for prion disease.
Around that time, Ionis discovered that antisense oligonucleotides, or ASOs — short strands of DNA, RNA or analogs that bind to target genes to modulate expression — could potentially treat prion disease. They found that ASOs could reach the brain when delivered at high doses via cerebrospinal fluid, or CSF, injection every few months, leading the company to explore ASOs for a range of neurological conditions.
The collaboration brought together Ionis’ clinical trial capabilities and the couple’s expertise in prion biology, launching early discussions about what it would take to develop a PrP-lowering therapy.
“The neuroscientist (at Ionis) gave us a checklist: a humanized mouse model, biomarkers (to monitor patients), a patient registry,” Vallabh said. “That became our to-do list for the next five years.”
In collaboration with scientists at Ionis, the National Institutes of Health, or NIH, and others, Vallabh and Minikel published a series of papers evaluating PrP knockdown by ASO as a therapeutic strategy for prion disease.
To evaluate ASOs for prion disease, Ionis scientists screened hundreds of DNA-like ASO candidates in cell culture to identify those that bind PRNP mRNA and trigger RNase H–mediated cleavage, thereby lowering PrP levels. They passed top hits to Vallabh and Minikel for disease studies.
In mice, ASO injections reduced both PrP mRNA and protein in the brain without obvious side effects. Most importantly, ASO-mediated PrP lowering extended survival in mice infected with mouse-adapted prions. Treating before or during early disease tripled lifespan of mice compared with untreated controls. Even when administered after symptom onset, ASOs provided some benefit.
These mouse experiments were the first time Vallabh and Minikel had seen live animals succumb to prion disease.
“Even though they're mice and we're people, it was still spooky how much the disease reminded us of (Vallabh’s) mom's illness,” Minkel said.
Vallabh said seeing the treated mice continue to live normally made her think, “I want to be those mice.”
After Vallabh and Minikel’s initial preclinical studies, Ionis continued optimizing the ASO strategy to target human PRNP, eventually selecting the ASO formulation ION717 to move into clinical trials.
Inside the clinical trial
Now that the ION717 trial is ongoing, no new patients can be recruited. Vallabh said she is happy it is underway, but “(i)t’s agonizing to be so much further down the development arc but still be telling people as of right now, there’s nothing we can do (to help them).”
During the trial, 56 prion disease patients will spend the first 30 weeks in a double‐blind, placebo‐controlled phase; afterward, all participants receive the ASO for 70 weeks, followed by 32 weeks of monitoring. Although the primary goal is to assess safety rather than efficacy, scientists will track disease progression and biomarkers, such as PrP levels in cerebrospinal fluid.
“A lot of outcomes are possible from this trial.” Vallabh said. “It would be positive to see any movement in the right direction. Markers (like PrP levels) coming down or rising at a slower rate even if (the changes) don’t rise to clinical benefit, could still tell us we are going in the right direction.”
If the treatment is safe and appears to slow the disease course, the next steps are easy: move on to the next trial phase. Otherwise, Vallabh and Minikel will go back to the drawing board.
There could be many reasons. “Lowering PrP could be the right idea, and yet the trial could still fail for any number of reasons” Minkel said. “Drug too toxic, not potent enough, dosed too high or too low or not frequently enough, not enough drug in the deepest brain regions, patients already too advanced … and so on.”
Vallabh said it is difficult to derive answers from neurodegeneration trials because they don’t know at what point a patient reaches the point of no return, or a milestone after which treatment will not be effective.
“It breaks my heart that in neurodegeneration we’re seeing so much money invested and still we can get to the end of a clinical trial and maybe not have learned one way or the other, because you can always argue that you treated too late. It’s something the field more broadly needs to get its head around.”
Because treatments work best when begun early, Vallabh and Minikel hope raising prion disease awareness will lead to earlier diagnoses. An accurate test exists — it measures a patient’s cerebrospinal fluid for prion-induced misfolding — but because prion disease is so rare, many doctors don’t consider it.
“Just in the last decade there’s been an explosion of ways to target DNA and RNA,” Minikel said. The couple is working on other ways to knock down PrP levels, including base editing, splice site alteration and epigenetic approaches.
“Whatever happens to this exact drug in this exact trial,” Minkel said, “some kind of first-in-human test like this of a PrP-lowering drug was always going to be the first step towards eventually developing a meaningful therapy. In that sense, whatever the outcome, it will be a step forward.”
Vallabh added: “Having something in (clinical trials) is not the finish line, it’s the starting line.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.