Proteins and lipids — a complicated relationship?


Researchers have been discussing for many years the role of the lipid matrix in regulating the activity and the organization of membrane proteins. A variety of effects have been singled out and studied qualitatively and quantitatively in model systems. However, the applicability of those results to living cells is — in many cases — unsatisfactory. Here, we would like to make the point that the complexity of the lipid–protein matrix in cells alters the physico-chemical mechanisms of protein–lipid interactions to an unknown extent when compared to model systems.
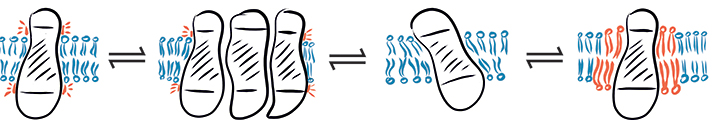 In a complex environment, hydrophobic mismatch may cause a protein to fluctuate between different states: (from left to right) hydrophobic mismatch, protein aggregation, protein tilting and recruitment of long-chain lipids.SCHEMATIC PROVIDED BY EVA SEVCSIK AND GERHARD J . SCHÜTZ
In a complex environment, hydrophobic mismatch may cause a protein to fluctuate between different states: (from left to right) hydrophobic mismatch, protein aggregation, protein tilting and recruitment of long-chain lipids.SCHEMATIC PROVIDED BY EVA SEVCSIK AND GERHARD J . SCHÜTZ
We shall discriminate between global and local mechanisms. Global mechanisms are mediated by lipid-bilayer properties; local mechanisms denote a direct molecular interaction between a protein and a lipid molecule. Examples of global effects include curvature, hydrophobic mismatch and preferential partitioning in phase-separated membranes (“rafts”) (1–3); examples of local mechanisms are the direct binding of cholesterol to CRAC domains (4) or of phosphatidylinositol 4,5-bisphosphate to protein subunits (5).
Formally, we may characterize a protein via its chemical potential µ, with the values for two different functional or structural states, µ1 and µ2. If 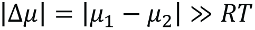 (R is the gas constant, T the temperature), a pronounced preference for one of the two states will occur. In contrast, for
(R is the gas constant, T the temperature), a pronounced preference for one of the two states will occur. In contrast, for  , we expect no preferred state.
, we expect no preferred state.
Most of the data for global mechanisms come from studies on simple, well-defined model systems, which allow for specifically addressing individual parameters. To emphasize the effects, such model systems usually are selected to achieve substantial contrast between µ1 and µ2. Examples would be the partitioning of proteins into ordered versus disordered phases in phase-separated lipid bilayers (6) or the recruitment of proteins to lipid vesicles with different curvature (7). Also, for local mechanisms, chemical potentials are the appropriate means of quantitating a protein’s state: µ1 denotes the lipid-bound state, µ2 the unbound state, and 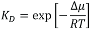 the equilibrium binding constant.
the equilibrium binding constant.
In cell membranes, however, a plethora of lipid species with varying properties, such as different head groups, acyl chain lengths and degrees of saturation, increases the complexity of the situation. The consequence for global mechanisms will be myriad chemical potentials describing the possible states of the protein, which can be approximated by a continuous energy landscape: Proteins essentially fluctuate between the different states. In some cases, cells may amplify the difference in chemical potential by de novo assembly of membrane structures, such as clathrin-coated pits, so that the partitioning or activity contrast will become more pronounced.
Also, in the case of local mechanisms, a variety of lipids may be able to interact with the protein of interest, potentially with only slightly different affinities. This leads to the recruitment of specific lipid species to the vicinity of the protein and an essentially continuous distribution of chemical potentials. Again, in some cases, preferred interactions of the protein with one type of lipid occur, yielding additional discrete values of µ.
What would be the consequences of a continuous distribution of chemical potentials? There would be no clear-cut states of a protein. For example, hydrophobic mismatch, on a stochastic basis, may lead transiently to demixing of the protein, the recruitment of a shell of long-chain lipids and membrane curvature. The system would fluctuate between these scenarios. Only in cases where the energy continuum splits up, or where distinct extra-states exist, can we expect distinct states of a protein. In conclusion, studies of well-defined model systems certainly help our understanding of fundamental physico-chemical properties, but the complexity of the live cell environment provides many more options to minimize the global energy of the system.
References
- Milovanovic, D. et al. Nat. Commun.6, 5984 (2015).
- McMahon, H.T. & Boucrot, E. J. Cell. Sci.128, 1065 – 1070 (2015).
- Van Meer, G. et al. Nat. Rev. Mol. Cell Biol.9, 112 – 124 (2008).
- Fantini, J. & Barrantes, F.J., Front. Physiol.4, 31 (2013).
- Hamilton, P.J. et al. Nat. Chem. Biol.10, 582 – 589 (2014).
- Dietrich, C. et al. Proc. Natl. Acad. Sci. USA98, 10642 – 10647 (2001).
- Hatzakis, N.S. et al. Nat. Chem. Biol.5, 835 – 841 (2009).
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.