Publication of 2,000 canine genomes provides toolkit for translational research

An international research consortium has generated and analysed 2,000 canine genomes. The resulting advanced genetics toolkit can now be used to answer complex biological questions, spanning dog domestication, genetic differences in breed morphology, behavior and disease susceptibility, as well as the evolution and structure of the genome. The study, published in the journal Genome Biology, describes the toolkit resource package and presents the first set of discoveries.
The publication is the culmination of efforts from the Dog10K consortium — 48 scientists across 25 institutions, contributing samples and resources to the immense analytical effort.
Jennifer Meadows is an Uppsala University research scientist and was a lead co-author of the study. “The goal was to produce a resource the global community could access, and which they could use to speed the translation of their own research, be that in the study of the shared ancestors of dogs and wolves, or the clinical treatment of cancers. All these avenues are exciting, and all can benefit from the Dog10K catalogue,” Meadows said.
The power of the Dog10K analyses lies in the depth of genetic diversity the team was able to capture. Canine samples were drawn from more than 320 of the approximately 400 recognised pedigree dog breeds, as well as niche populations of village dogs, wolves and coyotes.
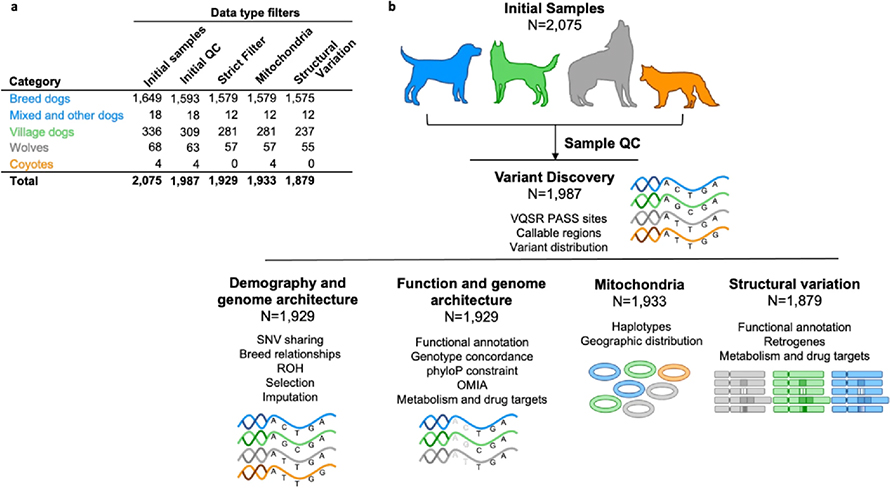
The team developed:
- A comprehensive catalogue of single nucleotide variants, including those which can disrupt protein coding genes and may influence the extremes of dog appearance and physiology. All pedigree dogs in the study were free from disease when sampled, suggesting that some of the variants that cause a loss of protein function do not cause obvious disease in that population.
- A catalogue of structural variants. This category of variants has a minimum size of 50 base pairs, and so impacts more space in the genome than a single nucleotide variant. Some structural variants were found to impact protein coding genes. This could mean that too much, or too little of the protein is made in that individual, but again did not cause obvious disease in the pedigree population.
- For both single nucleotide and structural variants, the Dog10K team illustrated how these variants are distributed within and between pedigree dog breeds, and how these variants may impact the study of potential drug therapies.
- Publicly available data processing pipelines. Combining the results from different studies can be made more difficult when different settings are used to map sequencing data, and then to call variation. The pipeline used by the Dog10K consortium is open to the community so that new or existing data can be processed and easily combined with this set.
- An imputation panel from the single nucleotide variants. Imputation is a process that allows researchers to infer variation at sites that they did not directly measure. The Dog10K team showed that by using the imputation panel, a dataset that contained 170 thousand single nucleotide sites, commonly used in genotyping array studies, can be expanded to up to 8 million sites. This allows researchers to reuse their valuable samples, and to revisit complex questions that need more data.
“We have just scratched the surface of the data’s potential,” Meadows said. “There is yet more genetic diversity left to be found in dogs, wolves and coyotes, but the Dog10K team looks forward to seeing how this first effort is applied by the canine science community.”
This article was first published by Uppsala University. Read the original.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.