From the journals: MCP

Analysis of large samples of proteins just got an upgrade. Read about the latest breakthroughs in proteomics in these papers from the journal Molecular & Cellular Proteomics.
Two approaches are better than one
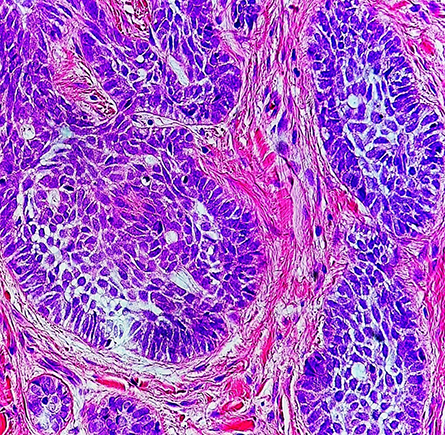
signatures in skin cancer cells like those shown in this high-magnification
micrograph. The cancerous cells are a darker purple than the surrounding stroma.
Proteomics studies sets of proteins in a system such as a skin cell. Researchers need to understand what proteins make up a given organism or cell type to determine how a system functions and how to distinguish one system from another. One step beyond looking at the genome, which is largely static in a given system, proteomics shines a light onto changing protein dynamics.
Researchers can compare the proteomes of healthy and cancerous skin cells to see what proteins are changed in cancer, for example, and then predict what pathways are being up- or downregulated as well as what proteins are indicators of a cancerous cell.
While the possibilities of proteomics research are endless, the current technology is a hindrance. Ina recent study in Molecular & Cellular Proteomics, Claudia Ctortecka and a team at the Vienna BioCenter in Austria combined two well-used analytical strategies in proteomics.
In the first, data-independent acquisition, or DIA, they fragmented all the peptides in a certain mass window and then analyzed the data. This differs from traditional data-dependent acquisition, or DDA, which isolates each peptide and then fragments each separately. Ctortecka and collaborators chose DIA because it provides more robust and similar quantification every time for the same sample and because DDA has problems merging large numbers of proteomes.
The authors combined DIA with tandem mass tag, or TMT, multiplexing, which allows researchers to run multiple samples at once by labeling each sample with different stable isotopes. TMT increases throughput of proteomics runs and the abundance of fragment ions available for peptide identification, but importantly, TMT by itself has problems with missing data points across multiple analytic runs. The researchers provide compelling data that combining DIA analysis with TMT experiments provides highly reproducible, quantitative proteome signatures that can be used to identify cell types and single protein knockouts.
A little sample goes a long way
One way to improve proteomics research is in the analysis tools. Another equally important aspect is initial sample processing. In some instances, rare cell phenotypes can occur, such as during the short mitosis cell phase. Researchers must be able to understand these short-lived cell phases, and they are difficult to analyze with traditional proteomics methods, which require lots of cells.
One method for studying rare cell phenotypes with low cell yield is fixing with formaldehyde to minimize perturbations to physiological processes. However, this fixative causes cross-linking between proteins, requiring additional digestion or other steps that could compromise the samples.
In arecent paper in Molecular & Cellular Proteomics, Van Kelly and a team at the University of Edinburgh describe a new processing method of digesting low-yield samples with protease. The researchers were able successfully to characterize proteome changes across 16 cell cycle states from TK6 cells. This technique could revolutionize the way proteomics addresses rare cell phenotypes and make better use of low yield samples.
Harnessing proteomics to find biomarkers
Proteomics has revolutionized the real-world impact of research on identifying and treating diseases. NeserinAli and a team at Lund University use their understanding of proteomics techniques to analyze acute differences between osteoarthritis disease stages.
Early-stage osteoarthritis is a degenerative joint disease affecting 5% of people between the ages of 35 and 54. Known biomarkers for osteoarthritis lack sensitivity and are not useful in tracking progression of the disease. Studies of the osteoarthritis proteome and proteomic analysis have been hindered by low availability of osteoarthritis biological samples, as sample collection procedures are rather invasive.
In their recent paper in Molecular & Cellular Proteomics, Ali and collaborators describe using state-of-the-art mass spectrometry to compare early- and late-stage osteoarthritis. Their work has led to key discoveries in potential pathways that osteoarthritis is using, both by identifying the proteins upregulated in osteoarthritis and by finding new biomarkers for osteoarthritis. The researchers also found biomarkers specifically for early-stage osteoarthritis, noting that this stage of the disease looks like a “raging battlefield” of proteins, while late-stage osteoarthritis is more like the “aftermath.” This work demonstrates how proteomics research can have an impact on disease diagnosis and treatment just by showing what proteins are involved.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.

Flipping lipids and slime molds
A dull first job nearly pushed JBC associate editor Todd Graham out of science. Then a slime mold project changed his path. Now, he studies membrane biology and reflects on discovery, persistence and mentoring through uncertainty.

How smelling death alters worm behavior
Researchers have found that the roundworm C. elegans can smell death, and it changes how the worms behave, reproduce and age.

A chance encounter with the lab
Payton Stevens never planned to become a pancreatic cancer researcher. A temporary job set him on a path from rural Kentucky to leading research on Wnt signaling and metastasis, where he now pairs discovery with mentorship and science advocacy.

Light-activated small molecule could transform eye infection treatment
Contact lenses raise the risk of infectious keratitis, a leading cause of blindness worldwide. A biotech company is commercializing a light-activated therapy using a ROS-generating molecule to rapidly kill microbes in the cornea to preserve vision.