Not so silent after all?

Craig Kaplan, an associate professor at the University of Pittsburgh, first saw the provocative claim on Twitter. A colleague had posted it with the succinct commentary, “Whoa.”
“I retweeted that with an additional “Whoa,” Kaplan said. “And then I immediately went to read it. … It’s such an unexpected result.”
The June Nature paper reported that nearly all synonymous mutations, changes in DNA that do not affect the amino acid sequence of the encoded protein, can reduce a yeast strain’s fitness, by 1% or 2% on average. It was a surprising finding from the lab of well-known evolutionary biologist Jianzhi (George) Zhang and his team at the University of Michigan.

But Kaplan, who studies transcriptional control, came across a technical shortcoming in the paper that concerned him. “I was really surprised to see such a fundamental error in experimental design — like, a basic, basic error in this type of experiment,” he said.
The study, which was inspired by an emerging recognition that synonymous mutations can do more than we’ve historically given them credit for, has sparked significant debate. Some scientists are concerned that it will make it harder for the community to accept future findings in this complex area of biology.
What can synonymous mutations do biochemically?
Most of our 20 amino acids are encoded by at least two, and sometimes as many as half a dozen, three-letter DNA codons. A synonymous mutation occurs when one codon is swapped for another that encodes the same amino acid. In textbooks, these are often called silent mutations: changes without impact.
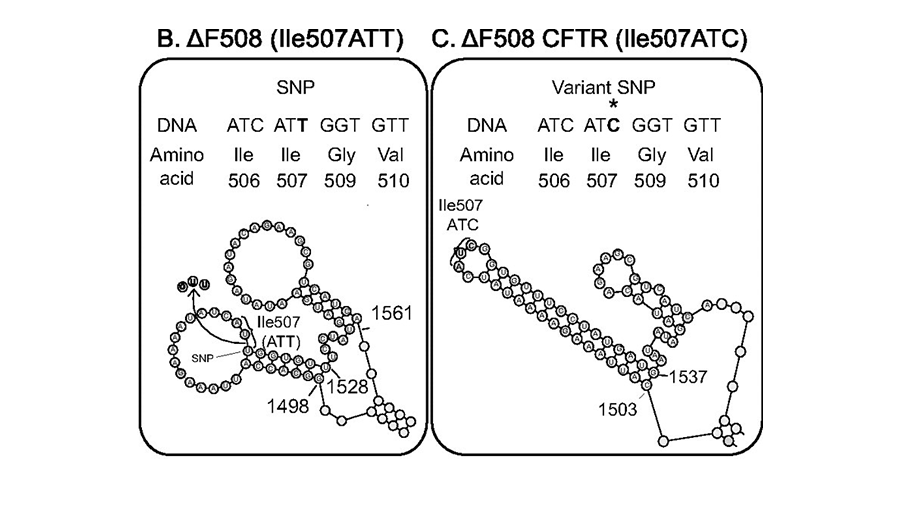
Synonymous codons are used at different frequencies in different species. Many biochemists are familiar with codon optimization, a technique to improve expression of a protein in bacteria, yeast or mammalian cells by shifting its codon sequence without altering the amino acid sequence.
“We’re living with a foot in two worlds,” said Patricia Clark, a biophysicist and professor at the University of Notre Dame who has studied the impacts of synonymous codon use in protein folding. “On the one hand, we know synonymous codons have effects. That’s why we change them to alter gene expression. But, on the other hand, we like to think these effects are rare and benign,” primarily cropping up when a gene from one species is expressed in another.
In recent decades, researchers have come to appreciate that codons’ biochemical effects may not be so rare. Ariel Bazzini, an associate investigator at the Stowers Institute for Medical Research, said, “They are silent from the amino acid point of view — but not from the regulatory point of view.”
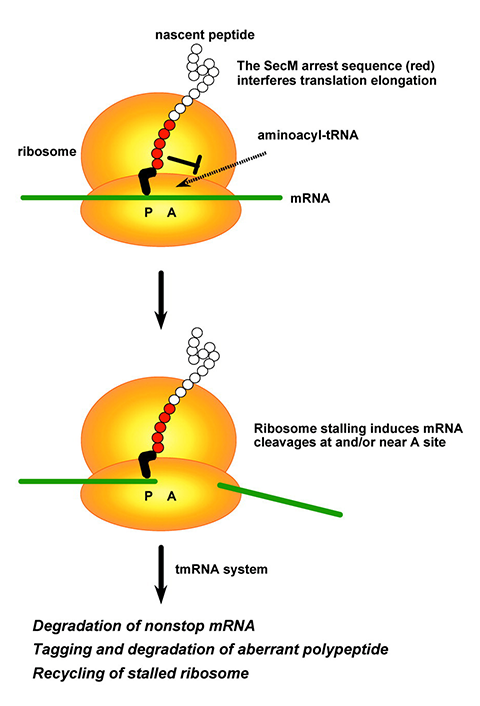
Mutations that change one codon to another can affect transcription by altering chromatin structure or promoter binding and might disrupt splicing sites. They may change the structure, and therefore the stability, of messenger RNA; stronger hairpins may stall the ribosome; and, conversely, less-stable secondary structure near the translation start site can improve ribosome loading. They also may alter the rate of decoding. Two codons for the same amino acid might be recognized by tRNAs with different abundance, subtly changing the speed at which the ribosome can progress. A ribosome’s speed can affect mRNA half-life: A stalling-prone transcript is more likely to be degraded. It can also alter co-translational protein folding; in 2020, Clark’s lab showed that shuffling the locations of comparatively rare and common codons disrupted folding of an antibiotic-resistance protein enough to change bacterial survival.
However, the extent to which each of those mechanisms affect the organism overall remains unclear, in part because it is difficult to study any of them in isolation. “The literature is filled with examples of uncontrolled variables — other changes that could be occurring that could be confounding the results,” Clark said. “In a cell, it’s very difficult to separate out all the potential mechanisms that could be affected by synonymous codon changes.”
What do synonymous mutations do to fitness?
Zhang became interested in synonymous mutation from an evolutionary perspective. If, as dogma once held, synonymous codons change nothing, then why are they used at different rates in different species?
Natural selection has had a long time to polish protein sequences. Therefore, most possible changes to a protein’s amino acid sequence — missense mutations, which alter a single amino acid, and nonsense mutations, which introduce a premature stop codon — compromise its function. It is widely accepted that nonsynonymous mutations tend to be harmful to an organism’s fitness; synonymous mutations are more often considered neutral.
Zhang was struck by two papers in 2018 that found similar fractions of synonymous and nonsynonymous polymorphisms that altered the rate of cell growth in yeast strains. That was when Zhang felt, he said, “We really have to check this at a large scale — and not just the polymorphisms, because polymorphisms are mutations that have been selected. How about de novo mutations?”
He and graduate student Xukang Shen set out to measure the impact of thousands of synonymous mutations on haploid yeast. They selected a group of genes that affect growth but are not essential to survival. Then, using CRISPR, Shen removed sections of about 150 base pairs from each gene, and he then added back to the resulting deletion strains a library of replacements that contained the missing DNA altered by just a single base pair each.
After generating hundreds of mutants — some synonymous, some missense and some nonsense — at each genetic locus, Shen grew all of them in a single flask with a wild-type strain and used high-throughput sequencing to determine which strains were present and how abundant they were. The strains’ ability to outcompete their relatives showed which were the fittest.
For most genes, they found, synonymous mutations could reduce a strain’s competitive edge.
“Many of their synonymous mutations caused a significant fitness defect — and that those effects were so big; those were both surprising,” said Clark.
Zhang argues that synonymous mutations, on average, are more costly than neutral. The lab collected some evidence linking reduced fitness to lower mRNA concentrations, though the correlation was not strong.
In a companion commentary, geneticist Nathaniel Sharp of the University of Wisconsin–Madison wrote that the paper showed “synonymous mutations are frequently just as harmful as the non-synonymous mutations that alter proteins, upending a common assumption about molecular evolution.”
The only problem was the technical shortcoming that Craig Kaplan was soon to observe: an Achilles heel that other researchers agree could invalidate the study’s conclusions altogether.

A methodological debate
Five weeks after the Nature paper was published, two preprints went online to rebut it. Kaplan coordinated and acted as co-corresponding author on one of them, with nine other molecular biologists as coauthors. They coordinated on Twitter, Kaplan said; they felt an imperative to move quickly because of how much attention the finding had received.
Besides the commentary in Nature, the paper got news coverage, including a piece in the New England Journal of Medicine. “There was a lot of speculation about how human geneticists and clinicians may need to pay attention to these types of variants… and that means it’s a really, really big deal to get this right,” Kaplan said.
The critique in Kaplan’s preprint is rather technical, but it boils down to a principle from introductory biology classes. “When you do an experiment, you want to minimize all possible variables, if you can,” Kaplan said. “At the same time you create your mutant pools, you should add back a wild type.”
The Zhang lab had used a wild-type strain that was edited twice, just as the mutants were. But instead of generating a wild-type rescue from each of the 21 deletion strains to use as a control, the lab compared all of them to a rescue strain derived from only one deletion mutant.
Kaplan argues that each deletion strain could have undergone any number of changes, such as off-target CRISPR effects, and transcriptomic, metabolomic, genetic or epigenetic compensations for the loss of an important gene. The experiment as designed would not have detected such changes, Kaplan said, and might have compared a mutant with several changes to a wild-type with just one.
Zhang agrees that it would have been best to design the experiment so that each mutant had a closely related rescue strain and to clone them all in one batch. However, he said, library-based cloning has technical limitations. “When you edit 450 mutants together, you can’t guarantee that you will get all 450,” he said. “This control is absolutely required for experiments, so we didn’t want to take that risk.”
The second preprint, a critique from a group of human geneticists, pointed out that among the 450,000 or more exomes stored in the U.K. Biobank, synonymous mutations were much less likely to be associated with disease than nonsynonymous mutations — indicating that fitness is at greater risk from amino acid changes than from changes to a transcript.
In a responding preprint of their own, Zhang and colleagues rebutted the geneticists on the grounds that their counterpoint compared all synonymous mutations with only those nonsynonymous mutations predicted to be harmful.
Ryan Dhindsa, a research fellow at Baylor College of Medicine, was one of the human geneticists who authored the critique. He said it’s true that one analysis, called a gene-based collapsing analysis, did draw from a dataset of potentially damaging missense mutants, but a second examined every observed variant, putatively harmful or not. He added that the rebuttal does not address the geneticists’ second point: that genetic studies of parents and their children have consistently shown much stronger links between nonsynonymous than synonymous mutations and disease.
“The expectation, supported by decades of evidence, is that most synonymous mutations will be neutral or nearly neutral,” he said. “Rarely, they can have a severe effect.”
Zhang’s lab responded to the molecular biologists by editing a group of wild-type rescues to complement the deleted genes with their original sequences. Although the median fitness of these strains restored to wild-type was lower than that of the original wild-type control, they argued, synonymous mutants’ fitness still averaged significantly lower.
“Whenever you report something that’s very contrary to what the sort of established dogma in the field is, the bar is much higher,” said Patricia Clark.

Where does that leave us?
At the moment, the field seems to be waiting to find out whether and when the preprint volleys from either side will be peer reviewed and published. “I think potentially people are just sort of holding their breath,” Kaplan said.
Zhang’s lab, meanwhile, is continuing to work on ways to replicate their work and confirm their findings more robustly. And Zhang remains cheerful about the debate. He said he has found the discussion to be “pretty standard,” adding, “I think it’s normal. I myself wrote a lot of critiques of other papers.”
Zhang said he sees his lab’s work as just one installment in a long conversation about what synonymous mutations do. He said that replication of the work using other genes and other organisms would bolster the field’s confidence that synonymous mutations are deleterious. Still, he believes that widespread acceptance will take time. “When you see something that is unexpected, it takes time for people to believe and also takes not just one study, but many studies.”
Bazzini remains skeptical that a single altered codon could produces changes of the magnitude the Zhang lab observed: “We know that synonymous mutations can affect RNA stability. But you need to have a lot.”
Clark worries that in the future researchers might write off new findings concerning synonymous mutations. “We've seen this happen in other research areas,” she wrote in an email. “A bold, broad claim that is later walked back could cause some people to dismiss the well-established effects that synonymous codons can have.”
Codons in biotechnology
Most biologists who aren’t directly researching synonymous mutations are familiar with the concept because of differences in codon usage between organisms.
Most species tune the amount of tRNA they produce to match the codons that are present (or vice versa).
An RNA that uses many codons that are rare in the organism that is expressing it may stall the ribosome frequently, harming protein yield and adding work to the protein-purification process.
Therefore, conventional wisdom is to optimize a plasmid by replacing its rarest codons with codons that are common in the target organism.
Several programs exist that will convert a plasmid between species to maintain robust RNA translation efficiency. iCodon and CHARMING, written in Bazzini’s and Clark’s labs, respectively, are two of them.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.

Flipping lipids and slime molds
A dull first job nearly pushed JBC associate editor Todd Graham out of science. Then a slime mold project changed his path. Now, he studies membrane biology and reflects on discovery, persistence and mentoring through uncertainty.

How smelling death alters worm behavior
Researchers have found that the roundworm C. elegans can smell death, and it changes how the worms behave, reproduce and age.

A chance encounter with the lab
Payton Stevens never planned to become a pancreatic cancer researcher. A temporary job set him on a path from rural Kentucky to leading research on Wnt signaling and metastasis, where he now pairs discovery with mentorship and science advocacy.