Sphingolipid metabolism: a potential therapeutic target in sickle red blood cells



About 300,000 babies around the world are born each year with sickle cell disease, a genetic disorder that affects about 100,000 Americans. The disease causes pain and inflammation due to blood flow blocked by sickle cells, resulting in serious complications such as strokes in childhood, acute chest syndrome, organ damage and premature death, with an average life expectancy of 45 years.
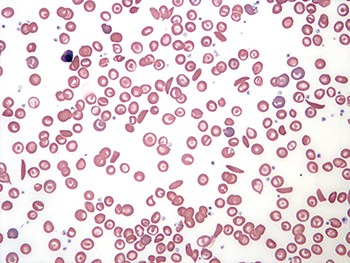
Sickle cell disease is caused by a point mutation in the gene for the beta subunit of hemoglobin. When red blood cells enter low-oxygen environments, the mutated hemoglobin polymerizes, leading to alteration of the cells’ shape. Sickle hemoglobin also has a high affinity for membranes, leading to accumulation of denatured sickle hemoglobin on the inner plasma membrane surface. This contributes to a wide range of dysfunctions of sickle red blood cells, including increased oxidative stress, increased surface phosphatidylserine exposure, increased adhesion to endothelium and hyper-production of extracellular vesicles.
While alterations of membrane shape and glycerophospholipid composition in sickle red blood cells have been analyzed extensively, the involvement of bioactive sphingolipids is less well studied. Sphingolipids such as sphingomyelin and ceramide are important structural components of the plasma membrane. They regulate the lateral domain structure of membranes, including formation of lipid rafts, which is critical for protein sorting and membrane signaling. Beyond structural functions, generation of ceramide by hydrolysis of sphingomyelin through the action of sphingomyelinase causes increased surface phosphatidylserine exposure on the cell surface, increased microparticle formation and increased adhesion of red blood cells to endothelium.
The bioactive sphingolipid that has received the most attention in the context of sickle cell disease is sphingosine 1-phosphate, or So1P. Red blood cells store So1P at a concentration of about 1 nanomole per milliliter of cells, which is higher than most cell types in the body. Red blood cell-derived So1P also can be released into the plasma, where it binds primarily to albumin and high-density lipoprotein. Once in the plasma, So1P regulates a range of physiological systems, most notably endothelial barrier integrity and the trafficking of lymphocytes and hematopoietic stem cells. New evidence suggests that So1P plays a role inside red blood cells as well. So1P may bind directly to hemoglobin, altering its affinity for oxygen and its affinity for the plasma membrane. Importantly, studies from our lab and others have shown that So1P is elevated in sickle red blood cells. This suggests new therapeutic opportunities for treatment of sickle cell-associated pathologies by manipulation of So1P production and metabolism.
Increased So1P in red blood cells could stem from changes in different metabolic pathways. Evidence from our laboratory and others suggests that sphingosine kinase activity is significantly elevated in sickle cell disease red blood cells. However, our studies and others show that sphingosine concentration also is elevated in these cells. Since the rate of So1P production is dependent on both the activity of sphingosine kinase and the availability of substrate sphingosine, the relative contributions of enzyme activity changes and substrate concentration to the increased red blood cell So1P are not clear. Further, it is unclear how red blood cell sphingosine is increased. One possibility is through increased ceramidase flux. Although we do not know whether the activity of ceramidase is affected in sickle red blood cells, our previous studies have demonstrated an increase in sphingomyelinase activity. This could increase the concentration of ceramide, which is the substrate for ceramidase. An additional possibility is that there is increased influx of sphingosine from the plasma, since our studies have shown increased plasma sphingosine concentration in sickle cell disease.
Most comprehensive studies of the red blood cell metabolome have ignored sphingolipids, and no comprehensive sphingolipid profile of red blood cells exists in the literature. In comprehensive proteomic studies of red blood cells, few sphingolipid-metabolizing enzymes have been reported. It is unclear whether this is because these enzymes are absent or because they are scarce. Additionally, the available literature on red blood cell sphingolipid metabolism is not entirely consistent. Some studies have identified activity of certain enzymes, while other studies have found none. The only enzyme consistently observed across multiple studies of red blood cell sphingolipid metabolism is sphingosine kinase. Given the inherent complexities of sphingolipid metabolism, an incomplete knowledge of the sphingolipid metabolism network in red blood cells may confound research efforts to exploit the role of So1P in sickle cell disease pathology.
The picture of sphingolipid metabolism in red blood cells that emerges includes multiple enzymes, fluxes and sphingolipid concentrations that likely are affected in sickle cell disease. The relative importance of these changes and the optimal therapeutic strategy for addressing them as a whole is difficult to determine.
Recent technological and methodological advancements may enable researchers to better understand the metabolism of bioactive lipids in sickle cell disease. First, lipidomics approaches using liquid chromatography–tandem mass spectrometry allow researchers to measure several lipids under steady-state and dynamic conditions. Second, computational systems biology allows researchers to integrate those measurements into a mathematical modeling framework, which allows for enhanced insight into system behavior and prediction of optimal therapeutic interventions.
Ultimately, we believe that the integration of lipidomics technology and systems biology can allow researchers to create a more coherent picture of the lipid dysregulation that underlies the many manifestations of sickle cell disease pathology.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.