JBC: A change in labs reveals a key to cataract formation

Researchers working to understand the biochemistry of cataract formation have made a surprising finding: A protein that was long believed to be inert has an important chemical function that protects the lens of the eye from cataracts.
 Though the labs where Eugene Serebryany did his graduate and postdoctoral research were just two stops apart on the subway, he had trouble replicating his experiments when he moved. Figuring out what had changed led to a discovery in cataract formation. Acela 2038/Wikipedia
Though the labs where Eugene Serebryany did his graduate and postdoctoral research were just two stops apart on the subway, he had trouble replicating his experiments when he moved. Figuring out what had changed led to a discovery in cataract formation. Acela 2038/Wikipedia
The lens is made up of cells packed with structural proteins called crystallins that form a dense gel. The gel’s optical properties — such as its transparency and the way it refracts light — help focus light onto the retina. But when crystallin proteins clump together, they scatter incoming light, forming cloudy deposits known as cataracts.
According to Harvard postdoctoral fellow Eugene Serebryany, lead author on a study in the Journal of Biological Chemistry, researchers long believed that crystallin proteins were chemically inert; except for aggregating over time, the proteins were not supposed to interact much with other proteins.
As a graduate student at the Massachusetts Institute of Technology, Serebryany used a mutant form of gamma-crystallin to mimic the damage ultraviolet light causes the protein. While studying how that mutation leads crystallin to clump up, Serebryany found something surprising: The mutant was more likely to aggregate if wild-type, or undamaged, protein was also present.
Eugene Shakhnovich of Harvard, who collaborated with Serebryany and his graduate adviser, Jonathan King, on the earlier studies, described the finding as “fairly striking.”
“If you had these damaged proteins in a test tube, they would not aggregate for a while,” Shakhnovich said. “If you had the wild-type protein, it would not aggregate forever. But then, when you mix the two, you see rapid and precipitous aggregation.”
Somehow, the healthy version of a protein everyone had thought was inert was causing a slightly damaged version to get much worse — and fast. Shakhnovich hired Serebryany at Harvard to continue studying how a supposedly inactive protein could cause this effect.
But Serebryany had trouble replicating the results of his own experiments. “It’s a different place, it’s a different set of instruments, a slightly different set of procedures. You see where this is going,” he said. “All of a sudden, experiments that were highly reproducible before were giving a lot of variability.”
In the Harvard lab, the wild-type crystallin sometimes caused mutant crystallin to aggregate, and sometimes it didn’t.
“Obviously, if there is suddenly variability, there is a hidden variable that we didn’t see before,” Serebryany said. He set up a series of experiments to try to pinpoint that variable.
Close comparison of the molecular weights of batches of the wild-type protein that caused or failed to cause the mutant to clump revealed a difference equivalent to the weight of two hydrogen atoms. This gave the researchers a hint that the protein’s redox state — whether two sulfur atoms were bound to one another or to hydrogen atoms — made a difference.
“By carrying out isotopically resolved mass spectrometry experiments, we got more than we bargained for,” Serebryany said. “Not only did the aggregation-prone mutant acquire one internal disulfide bond per molecule during the aggregation reaction, but the aggregation-promoting wild-type protein lost its disulfide at the same time.”
By mutating crystallin’s sulfur-containing cysteine amino acid residues one by one, Serebryany found that two residues close together on the surface of the protein acted as a kind of switch. When they bound, forming a disulfide bond, crystallin was able to push damaged molecules toward aggregation. When each was bound to a hydrogen atom, wild-type crystallin was inert.
How could one bond between amino acids on the surface of this protein make it drive other proteins to aggregate?
The team found that although the disulfide bond forms easily, it strains the protein’s structure. This made each molecule likely to pass along the disulfide bond to another nearby, receiving two protons in return. In this way, the disulfide bond could be passed constantly around among crystallin protein molecules like a hot potato.
Given a whole population of undamaged crystallin proteins, that could go on indefinitely. But if one protein was already a little damaged, the authors showed, it caught the hot potato with a different set of cysteines, which were less able to pass it on. This drove the damaged protein to aggregate.
According to Shakhnovich, the team is working on peptide treatments that might keep the hot potato from reaching damaged proteins. Serebryany hopes such peptides “could actually soak up some of those disulfides and delay the time that it takes to form the more aggregation-prone species.”
That could lead to slower cataract formation for patients.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Gaze into the proteomics crystal ball
The 15th International Symposium on Proteomics in the Life Sciences symposium will be held August 17–21 in Cambridge, Massachusetts.
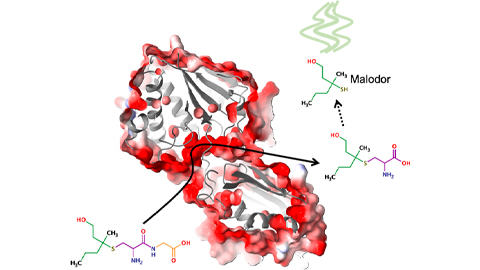
Bacterial enzyme catalyzes body odor compound formation
Researchers identify a skin-resident Staphylococcus hominis dipeptidase involved in creating sulfur-containing secretions. Read more about this recent Journal of Biological Chemistry paper.

Neurobiology of stress and substance use
MOSAIC scholar and proud Latino, Bryan Cruz of Scripps Research Institute studies the neurochemical origins of PTSD-related alcohol use using a multidisciplinary approach.
Pesticide disrupts neuronal potentiation
New research reveals how deltamethrin may disrupt brain development by altering the protein cargo of brain-derived extracellular vesicles. Read more about this recent Molecular & Cellular Proteomics article.

A look into the rice glycoproteome
Researchers mapped posttranslational modifications in Oryza sativa, revealing hundreds of alterations tied to key plant processes. Read more about this recent Molecular & Cellular Proteomics paper.
Proteomic variation in heart tissues
By tracking protein changes in stem cell–derived heart cells, researchers from Cedars-Sinai uncovered surprising diversity — including a potential new cell type — that could reshape how we study and treat heart disease.