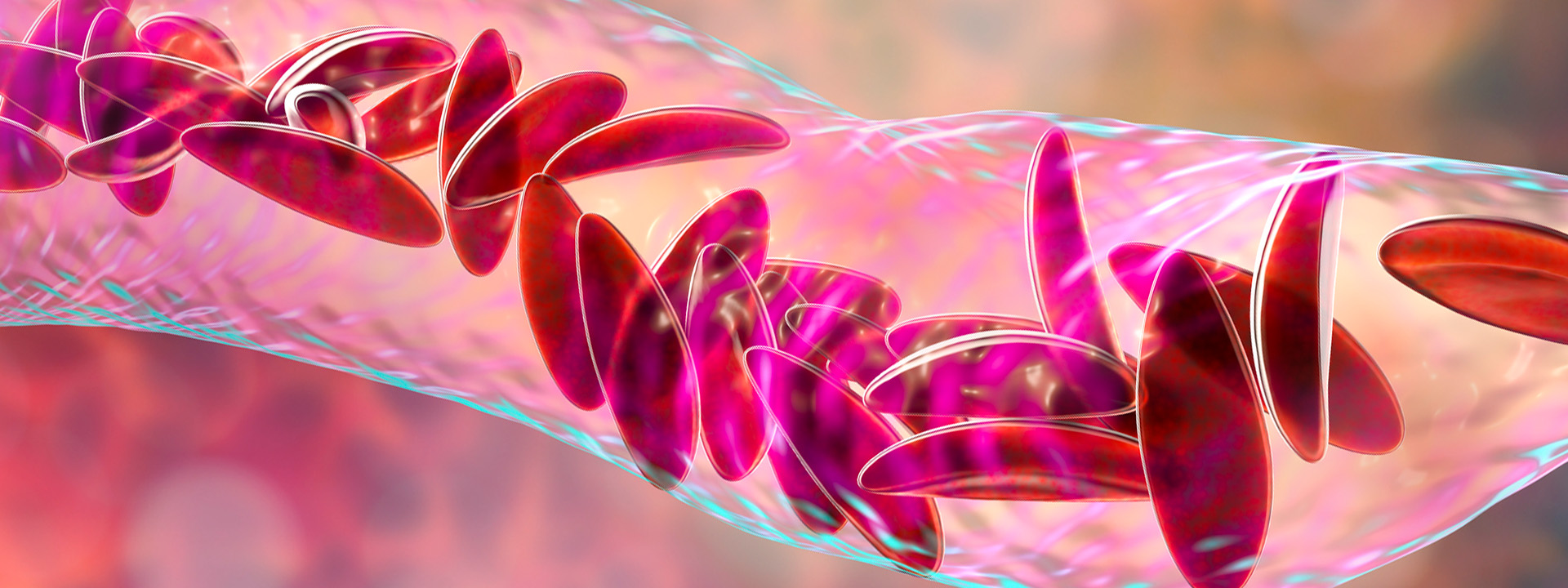
A road to survival

Te'Avionna Rowe, or Te’A as most people call her, is a bright 14-year-old and captain of her junior varsity cheerleading squad in Florida. She also has sickle cell disease.
“Mostly my days are pretty normal, like any other kid,” Te’A said. “When I’m in pain, that’s when it changes.”
A few months after Te’A’s mother, Raytoyia Brooks, gave birth, she received an ominous letter stating her baby’s blood might be abnormal. After a follow-up appointment, Te’A was diagnosed with sickle cell disease.
“I was completely devastated,” Brooks said.
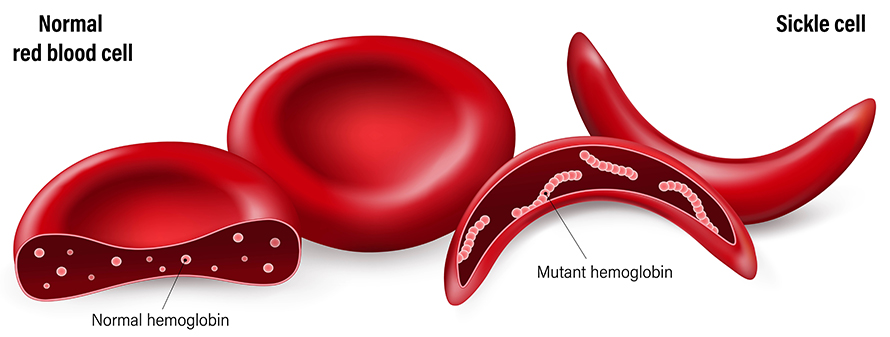
Sickle cell disease is a debilitating, genetic disorder that affects 100,000 individuals and 0.3% of Black or African-American births in the U.S. It causes normally disk-shaped red blood cells, which move easily through veins and arteries, to warp into a sickle shape.
The most common symptom is vaso-occlusive crises, when clumps of the crescent-shaped blood cells get stuck in vessels and block blood flow. These painful obstructions can lead to stroke, infection, eye problems and premature death.
The first six months of Te’A’s life were uneventful, and then she had her first crisis. This episode marked the beginning of a tough period.
“We were in the hospital all the time,” Brooks said, “sometimes every month.”
A traffic jam of red blood cells in her spleen caused Te’A’s early crises. The solution: blood transfusions every three to four weeks for six months to replenish her normal red blood cells. At the time, her only other therapy was penicillin with potassium to prevent infections.
For many years, the treatment Te’A received as an infant was the norm for sickle cell patients. Since her birth, however, several drugs have been developed and approved, and stem cell transplants have been found to be effective in some patients.

In December, the U.S. Food and Drug Administration approved two gene therapies. One of these, Casgevy, is a CRISPR–Cas9 gene therapy from Vertex Pharmaceuticals and CRISPR Therapeutics, the latter co-founded by Nobel laureate Emmanuelle Charpentier. The FDA also approved bluebird bio’s lovo-cel for sickle cell disease — a lentiviral-based gene therapy. Casgevy was approved in the United Kingdom in November.
However, even these potentially curative treatments require chemotherapy that can have devastating and lasting side effects, so researchers press on in the quest for a safe and effective cure.
A new lease on life?
In 2019, Victoria Gray, a mother of four from Mississippi, became the first sickle cell patient to receive Casgevy at age 34. Before the treatment, Gray had to rush to the emergency room at least once a month and was often hospitalized for weeks at a time, she said; during one particularly bad period, she spent her children’s birthdays, Thanksgiving and Christmas in the hospital.
“I wasn’t living; I was just existing,” Gray said. “I would just wake up and go from the bed to the couch.”

Gray said she decided to pursue treatment options outside of FDA-approved therapies when her son began misbehaving at school.
“His behavior had changed because he thought I was going to die,” she said. “So, I knew that I had to fight to survive for my kids. It wasn't just about me. I want to live for them.”
Since receiving Casgevy, Gray said she has a “new lease on life” and her disease no longer gets in the way of working as a cashier at Walmart and caring for herself and her family.
Patients and carriers

As director of the Sickle Cell Disease Program at the Johns Hopkins All Children’s Cancer & Blood Disorders Institute in Florida, Tamara New has seen how the disease gets in the way of patients living their lives. New oversees the care and education of Te’A and hundreds of other children. She also helps her patients become advocates for themselves as they move from pediatric to adult care.
“It's worse than having a kidney stone and worse than a woman in labor because it can be all over,” she said of the pain individuals with sickle cell disease experience. “The vast majority of my patients … come to see me for their appointments; they take their medications; and they do their very best. But it is a very unforgiving disease. And many of my patients will suffer from complications despite doing all the right things.”
Sickle cell disease, the most common genetic disorder in humans, is caused by a mutation in the hemoglobin beta globin chain gene, which then causes beta-globin subunits of hemoglobin to stick together. The mutant gene is recessive; an individual needs two copies, one from each parent, to be affected.
Though Brooks knew she carried the trait, Te’A’s father was not aware that he did.
“Her father thought I did something wrong during my pregnancy,” Brooks said.
In countries where the disease is more common, people often find out their carrier status at a young age. Nigeria is the most sickle cell endemic country in sub-Saharan Africa with approximately 2% to 3% of the total population affected. About 25% of Nigerians carry the trait, compared with just 7% of African Americans in the U.S.

Oluwaseyefunmi Adeniran grew up in Nigeria and now teaches biochemistry at Sefako Makgatho Health Sciences University in the Republic of South Africa. She found out she was a carrier when she was eight years old and was educated about how the disease is passed down.
“It has been drummed into my mind that, as a carrier, I must not marry someone who has the same genotype,” Adeniran said.
The disease carries little stigma in Nigeria and West Africa, she added; it is discussed openly within and outside of health care settings.
“Before a lot of my contemporaries go into a relationship, they are aware of each other’s genotype,” Adeniran said. “Once you start developing romantic feelings for anybody, you ask for their genotype. If it’s not a match, you do not let the relationship progress.”
A drug success — and side effects
Before the two gene therapies were approved in December, just four effective pharmaceuticals were available to sickle cell patients. None is curative, and only one, hydroxyurea, significantly reduces mortality, the frequency of painful crises and the need for blood transfusions.
Most physicians suggest patients with sickle cell disease take hydroxyurea daily.

Griffin Rodgers is one of the researchers who discovered the therapeutic benefits of the drug. Now the director of the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health, Rodgers has studied hemoglobinopathies for more than 40 years. He became interested in hematology when three of his friends died of sickle cell disease before they graduated from college.
“I wanted to see if I could break some ground in this area to help improve the lives of people affected with sickle cell disease and related disorders,” he said.
After Rodgers finished his medical training in 1982, he began researching sickle hemoglobin with Alan Schechter, a senior investigator at the NIDDK.
He likened the aggregation of sickle hemoglobin molecules inside a red blood cell to a “reinforced steel pipe,” which causes the cell to lose flexibility.
Adult hemoglobin has four components, two alpha and two beta globin subunits, while fetal hemoglobin is made up of alpha and gamma globin. Most babies with sickle cell do not exhibit symptoms until they are about six to nine months old, Rodgers said, because fetal hemoglobin persists in their bloodstream and does not cause obstructions. As infants age, fetal hemoglobin is replaced by adult hemoglobin and with it the mutant beta globin protein. Sickle cell patients who express abnormally high amounts of fetal hemoglobin into adulthood often show little to no disease.
Based on these data, Rodgers and Schechter hypothesized that fetal hemoglobin might hold therapeutic potential.
“We thought, if we could turn fetal hemoglobin back on, that might inhibit this sickle hemoglobin polymerization and thereby change the course of disease,” Rodgers said.
In a 1990 study, Rodgers and Schechter showed that hydroxyurea increased fetal hemoglobin levels and decreased destruction of red blood cells in 70% of sickle cell patients. A follow-up study in 1995 showed the drug also decreased crises and the need for blood transfusions. The FDA approved hydroxyurea for sickle cell disease to treat adults in 1998 and children in 2017.
“Thinking back now, had this drug been available when I was in high school, I still might have my friends to talk to today,” Rodgers said.
Hydroxyurea is generally affordable, New said, but some patients and their families choose not to use it because it is a chemotherapeutic drug and can occasionally cause side effects, including abdominal pain, discolored nail beds and hair thinning.

Brooks hesitated to put Te’A on the drug.
“I kept her off of hydroxyurea for about a year or so because of the chemo aspects of it,” Brooks said. “Then, her blood numbers kept coming back all wacky. The doctors told me the drug would really help stabilize her. So, eventually, I didn't really have a choice but to go ahead and put her on it. I just want what is best for her.”
Since Te’A started taking hydroxyurea, she has far fewer emergency room visits, Brooks said.
According to a study published in the journal Blood Advances, 30% of adult patients who are prescribed hydroxyurea choose not to take it for fear of adverse effects.
“The community is advocating for medications outside of the chemotherapy family,” New said.
Infertility and inequities
Until late last year, the only potential cure available to sickle cell patients was a hematopoietic stem cell transplant. In children, this replaces the abnormal stem cells in bone marrow with healthy cells from an eligible brother or sister. Only a sibling can donate hematopoietic stem cells to a pediatric patient. However, scientists are evaluating the safety and efficacy of a transplant from a half-sibling, parent or unrelated donor.
Before receiving a stem cell transplant, patients must undergo myeloablative therapy, a treatment with harsh chemotherapeutic drugs and radiation to eliminate their own bone marrow stem cells to make room for the transplanted cells. Due to the havoc that sickle cell has wreaked on adult patients’ vital organs throughout life, this regimen is risky, Rodgers said, so adults are rarely considered for a transplant.
Also, though a stem cell transplant may offer a cure, the myeloablative chemotherapy that often precedes it causes infertility in more than 80% of patients.

Sophie Lanzkron, director of the Sickle Cell Center for Adults at Johns Hopkins Hospital in Baltimore, oversees the treatment of over 600 patients.
“We know these (myeloablative) drugs absolutely cause infertility, both in men and women,” Lanzkron said.
Furthermore, men and women with sickle cell disease usually have lower baseline fertility than the general population, and medical insurance rarely, if ever, covers fertility preservation for these patients.
“If you're a kid with cancer, your fertility preservation is paid for,” Lanzkron said. “If you’re a kid or adult with sickle cell disease, it's not. We need to fix this problem.”
Some researchers are working to devise nonmyeloablative transplant strategies that use less toxic chemotherapy and/or radiation, which will do less damage to reproductive organs. Rodgers has successfully treated more than 60 adults with nonmyeloablative hematopoietic stem cell transplants using sibling donors.
Lanzkron blames structural racism within the medical system for the inequities faced by sickle cell patients. The disease receives more than three times less federal research funding and 75 times less foundation funding than other genetic diseases, such as cystic fibrosis, which primarily affects white individuals.
“It’s not rocket science,” she said. “We need to make sure people have access to care and understand their disease.”
Gray has experienced racism within the medical system firsthand, she said. She sometimes hesitated to visit the emergency room during her crises because providers would refuse to administer adequate care.
“I’ve had doctors tell me that I was just addicted to drugs and that my pain isn’t real,” Gray said. “You just get tired of trying to explain your pain because it doesn’t show on the outside. (Doctors and nurses) would second guess it all the time.”
In the face of this discrimination, Gray said, people rarely advocated for her.
“I’ve even had nurses that wanted to say something” about the mistreatment, Gray said. “But they were afraid that speaking up would cost them their job.”
Gene therapy to the rescue
Casgevy and lovo-cel, approved in December by the FDA, are the first potentially curative therapies for sickle cell patients who are not eligible for traditional stem cell transplants.
Both therapies edit a patient’s own hematopoietic stem and progenitor cells, outside their body. After the cells are edited and the patient is conditioned with myeloablative drugs, physicians reintroduce the millions of edited cells. These progenitors expand and differentiate into white and red blood cells over several months. Unlike a traditional stem cell transplant, this strategy curbs the risk of the patient developing graft-versus-host disease, which occurs when the transplanted immune cells recognize the patient’s own tissues as foreign and attack them.
Casgevy and lovo-cel use different strategies to combat the disorder caused by the mutant beta globin gene.
Like hydroxyurea, Casgevy takes advantage of fetal hemoglobin’s properties.
Specifically, Casgevy uses CRISPR–Cas9 to target BCL11A, a repressor of the fetal hemoglobin gene, using a guide RNA. The precise target site is a residue within the BCL11A enhancer region, which, once modified by the Cas9 nuclease, takes the brakes off the fetal hemoglobin gene, allowing transcription and later translation to occur. Therefore, in addition to carrying sickle hemoglobin, patients who have received Casgevy also express high levels of functional fetal hemoglobin, which drowns out the sickle’s damaging effects.
In the latest safety and efficacy clinical trial, patients who received exa-cel sustained high levels of total hemoglobin, similar to what is seen in healthy adults. Most needed no blood transfusions and were free of painful crises for at least one year after treatment.
Gray, who volunteered to be the first patient with sickle cell disease to receive Casgevy, has had no emergency room visits, hospital stays or crises for more than four years.

“I feel cured,” Gray said. “I can do anything I want now. … My life has changed dramatically with just a leap of faith.”
Since receiving Casgevy, Gray said she has become an unofficial spokesperson for gene therapy and patient education.
“It has been a joy, and it has changed my outlook on sickle cell and life.”
The other recently approved therapy, lovo-cel, uses a lentiviral vector to introduce a modified beta globin gene into patient stem cells. This modified gene produces an antisickling hemoglobin protein, which is designed to inhibit the polymerization of mutant sickle hemoglobin, making it less likely to form blockages in the circulation.
After lovo-cel treatment, 96% of the 36 sickle cell patients enrolled in a safety and efficacy clinical trial were free of crises and, on average, showed effective DNA editing in over 80% of red blood cells for at least two years. Furthermore, patients sustain persistent expression of the antisickling hemoglobin for more than 5 ½ years after lovo-cel treatment.
Risks and costs
Gray, and many other people, are saying these gene therapies are cures for sickle cell disease patients. However, Lanzkron urges caution.
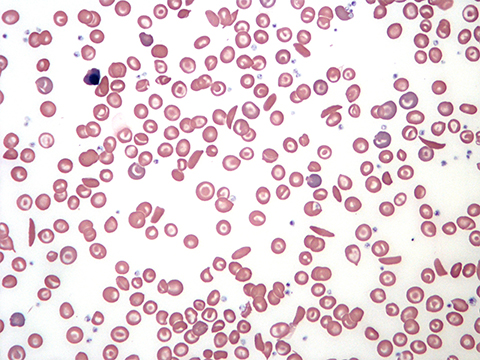
For a patient to be cured, Lanzkron said, they must be free not only of crises and the need for transfusions but of signs of hemolysis as well. Hemolysis, or the destruction of red blood cells, is a fundamental feature of sickle cell disease and underlies many symptoms.
“(After receiving gene therapy), these patients have ongoing hemolysis, and we know that is a big problem,” Lanzkron said. “These therapies aren’t truly curative, but I think they are transformational. We don’t know how long these effects last yet.”
Lanzkron also said she is concerned about the long-term effects of fetal hemoglobin after Casgevy treatment. According to a perspective published in the journal Blood, patients with high fetal hemoglobin levels can still exhibit severe disease.
“I've had patients with high levels of fetal hemoglobin who have had multisystem organ failure,” Lanzkron said. “(Gene therapies) seem to decrease vaso-occlusive episodes. I just think we have a lot more to learn about these therapies.”
Despite her reservations, Lanzkron said she would “absolutely” recommend gene therapy to her patients who could benefit from it. However, she emphasized that, before deciding on gene therapy, patients must consult with a sickle cell expert physician.
“Let us not forget what a horrible disease it is,” Lanzkron said. “We need these therapies. I will offer these therapies, and I will talk to my patients about them. But it's really a shared decision model with the limited amount of information that we have available.”
Of the five gene therapies approved by the FDA before December, most cost between $1 and $2 million. However, some can have price tags of up to $3.5 million.
According to the Institute for Clinical and Economic Review, Casgevy and lovo-cel could be priced at up to $1.93 million to be cost-effective, a measure that estimates how much it costs to gain a unit of a health outcome, such as a year of life gained or a death prevented.
In 2022, each patient with sickle cell disease incurred out-of-pocket medical costs totaling up to $44,000, with insurers covering approximately $1.7 million per patient. Economists predict that these new drugs could cost a single state Medicare program more than $30 million per year.
Many of her patients at All Children’s already incur thousands of dollars in medical expenses each year, New said, and a curative treatment may offer families a reprieve from lifelong medical costs.
However, Rodgers said gene therapies are not going to end sickle cell disease worldwide.
“The cost associated with these therapies with curative intent would be cost prohibitive, particularly if you needed to scale it to the 100,000 people that have the condition in this country and millions worldwide,” Rodgers said. “So we still need therapies like pills or small molecules that you would be able to effectively distribute and manufacture to change the health of all of these patients.”
On the horizon: one and done
Many research groups and companies are working to make gene therapies safer, more accessible and cheaper for patients around the world. Several of these initiatives aim to eliminate myeloablative conditioning from the treatment regimen.
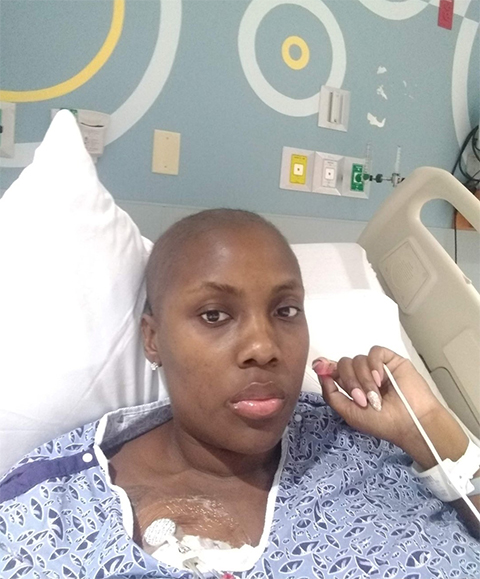
Gray said she developed mouth sores from the myeloablative chemotherapy while receiving Casgevy.
“I couldn't eat for over a week,” she said. “It was too painful. It even hurt to swallow water. I was extremely tired. But I was already tired with my sickle cell. So that wasn't really new to me.”

A team of researchers at the University of Pennsylvania, including Laura Breda and Osheiza Abdulmalik, showed that it is possible to genetically modify blood stem cells directly within the bodies of mice in what they call a “one-and-done” therapy. This could eliminate the need for currently used harsh myeloablative conditioning treatments.
Breda, a research assistant professor at the Children's Hospital of Philadelphia, said the researchers hope to develop treatments for genetic diseases, including sickle cell, available to patients in developing nations.
Abdulmalik is a research associate scientist at the same hospital. “The prevalence of sickle cell disease outside of the United States, in places like sub-Saharan Africa and India, is just not acceptable in the world we currently live in,” he said. “Therefore, some of us have started to focus our careers on trying to alleviate some of these inequities.”
Their gene editing approach uses lipid nanoparticles, or LNPs, and various cargoes to condition the bone marrow as well as deliver the gene therapy.
To make room for edited cells to expand, Breda and colleagues delivered an mRNA encoding a pro-apoptotic protein, p53 upregulated modulator of apoptosis, to hematopoietic stem cells using an LNP conjugated to CD117, a molecule that binds a receptor on hematopoietic stem cells. This binding can facilitate LNP update by the cell.
Their gene therapy approach uses a Cas9 fused to an adenine base editor and a single guide RNA, all encapsulated in an LNP conjugated to CD117 to edit diseased genomes. In their study, the team showed that these LNPs can efficiently edit sickle cell hematopoietic cells in a dish to reverse their sickling phenotype.
The team said this therapy would likely be more affordable and better tolerated by the patient and have fewer side effects than myeloablation with chemotherapy.
“This approach would circumvent all the issues with the traditional ex vivo gene editing approach,” Breda said.
More research will show whether the team can optimize and combine their conditioning and gene therapy strategies in a human to one day treat sickle cell disease.
Breda described their strategy as a “plug and play approach” that could easily be adapted to treat other genetic diseases.
Engineering an enzyme
Outside of academia, companies including Scribe Therapeutics are also developing gene therapies for diseases such as sickle cell.

After pursuing his Ph.D. in Jennifer Doudna and Dave Savage’s labs and a brief stint as an independent investigator, Benjamin Oakes cofounded Scribe Therapeutics in 2018 to use molecular engineering to bring CRISPR-based genetic medicines to the market.
“Scribe’s main focus is on how to bring genome editing in vivo and to larger patient groups, so that we all have access to this amazing technology,” Oakes said.

Their technology focuses on making the next generation of CRISPR–Cas safer and more efficient. During his Ph.D. studies, Oakes and colleagues described the CasX protein. At baseline, CasX does not edit the human genome well, Oakes said, but he saw this as an opportunity to mold it into a better genomic editor because this protein is naturally smaller, making it easier to deliver, and behaves differently than Cas9.
“Over the past four or five years, we have tested every single change you can imagine making to this enzyme,” Oakes said. “We have iterated again and again and again using protein evolution, RNA evolution and the coevolution of both together to do things like improve protein stability, DNA binding and DNA unwinding as well as improving the therapeutic characteristics of cleavage and ability to target a location of DNA and create a double-strand break.”
Oakes said Scribe’s version of CasX is more than 120 mutational steps away from the original protein. Their engineered enzyme can target a single nucleotide polymorphism on one allele.
“We’re really dramatically altering the landscape of how these enzymes look on both the protein and the RNA front,” Oakes said.
Scribe is partnering with the pharmaceutical company Sanofi to encapsulate their CasX gene therapy, composed of CasX and a programmable guide RNA, into targeted LNPs to make these genetic medicines easy to deliver.
Scribe’s gene therapies would not require a stem cell transplant and could be “undertaken anywhere in the world,” Oakes said. “It’s no more complicated than getting an infusion.”
Gray said she is thankful for all the scientists behind gene therapy.
“The long hours and sleepless nights put into these types of treatments really makes a difference,” she said. “So, keep doing what you are doing and keep researching.”
Patient takeaways
The FDA approved Casgevy and lovo-cel only for individuals 12 or older. However, future clinical trials may test the two gene therapies in pediatric patients.
New said gene therapy could be transformational for children with sickle cell disease.
“If gene therapy is able to lead to a lasting cure for patients, it is something that I think a lot of our families would be grateful to have,” she said. “I hope it opens up a cure for a lot more of our patients.”
However, the field still has a long road ahead to make sure this treatment is safe and effective for children, New said.
“It's not always easy to switch from a therapy that works well in adults to children. In adult medicine and clinical trials, they may be looking five to 10 years down the road. In pediatrics, we need to look 15, 20 or 25 years beyond treatment because, if I get rid of your sickle cell, I want to know that it's gone. I don't want it to suddenly come back when they are older.”
At 14, Te’A qualifies for the new gene therapies, but she and Brooks said they would only consider them if an approach came along that did not require myeloablation. Brooks said it would be amazing if Te’A could receive a potentially curative gene therapy.
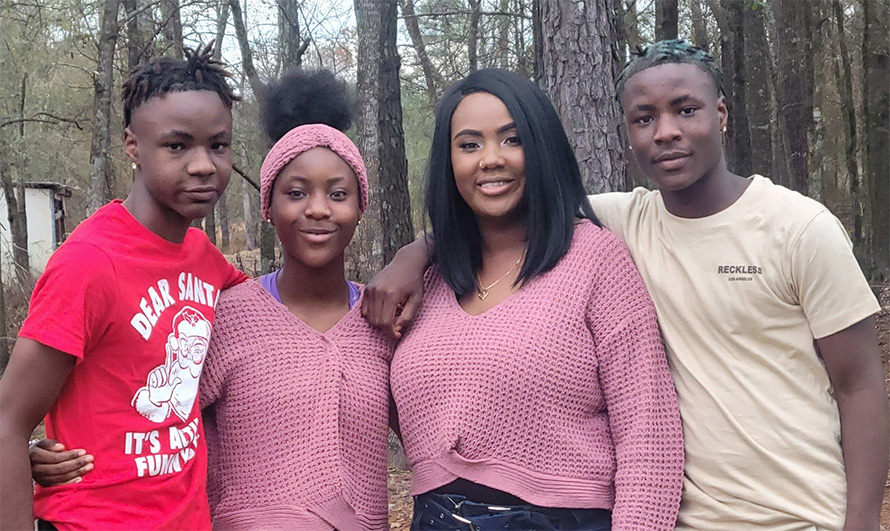
Gray said she has encouraged other patients to pursue gene therapy, and many have gone on to enroll in clinical trials.
“The opportunity for them to cut ties with the hospital is big,” Gray said, “because we do experience a lot of stigma when we go into the ER. So to be relieved of that, relieved of the pain, and able to get rid of our huge medicine baskets that we are tied to is life-changing.”
She added, “I think it is a fair trade: (temporarily) giving up your hair for the chance of having a full life.”
In Nigeria, Adeniran has lost friends and family to sickle cell disease when they were young, but she said she is optimistic because affected individuals in her community are living longer.
“People with sickle cell disease can have a healthy, fulfilled life,” Adeniran said. “It's not a death sentence. Even though we are looking for solutions, people with sickle cell go on to have long beautiful lives.”
Gray said she is happy to have helped open the door to gene therapy, and she advised all sickle cell patients to “hold on and don’t give up yet.”
“Change is coming. What once was science fiction is now fact.”
What's next?
To address the weaknesses and side effects of CRISPR-Cas9 therapies, researchers have developed two new methods for gene editing. Read about these recent developments in "New kids on the block: Base and prime editors."
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.