Today is International 15q Day

Nov. 15 is internationally recognized as a day to raise awareness for 15q neurodevelopmental disorders, which is an overarching categorization for three distinct conditions: Angelman syndrome, Dup15q syndrome and Prader–Willi syndrome. Each condition is unique, yet all are caused by a change or mutation in the 15th chromosome.
The National Library of Medicine estimates that Angelman syndrome affects one in every 20,000 people and Prader–Willi syndrome affects one in every 10,000 to 30,000. Dup15q has an unknown frequency, and it is estimated that it could be as high as one in every 5,000 people.
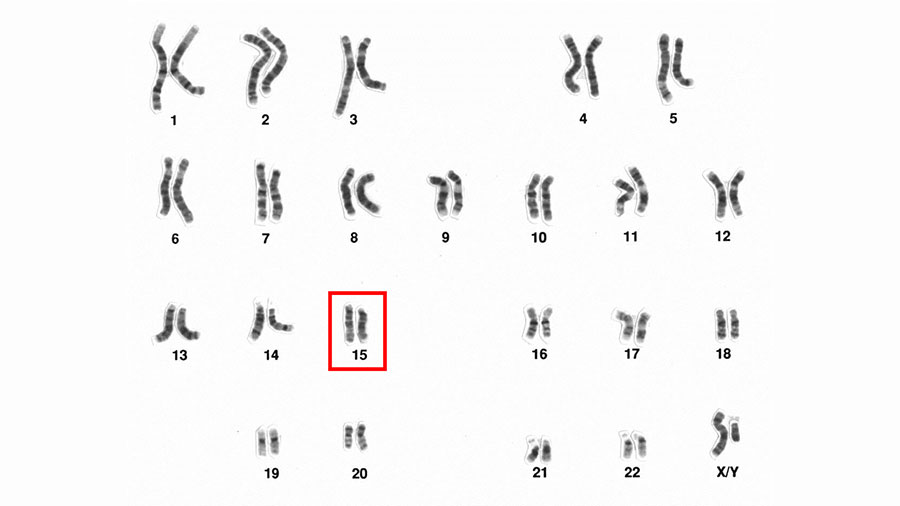
Prader–Willi Syndrome
Prader–Willi syndrome is caused by a genetic mutation of undefined mechanisms. Current research suggests that this mutation causes an inability to express paternal genes; this may be due to a child inheriting two maternal chromosomes (as opposed to one maternal and one paternal copy), a defect in the paternal gene that prevents proper expression, or a complete lack of paternal genes on the chromosome.
Typically, symptom presentation begins around 2 years of age, and disease presentation includes hyperphagia, poor responsiveness, underdevelopment and hypotonia. Hyperphagia often leads to obesity, which means that individuals with the syndrome maintain a higher risk of experiencing obesity-related complications such as Type II diabetes, high blood pressure, elevated cholesterol and heart disease. Additionally, decreased hormone production due to underdevelopment may lead to complications including sterility and osteoporosis.
The critical region for Prader–Willi Syndrome is located at chromosome 15(q11-13), in which exon 1 is a key region for imprinting. In PWS, exon 1 is maternally imprinted, meaning that paternal allele expression does not occur. Genetically, this can be caused by one of three things: deletion of a critical region, uniparental disomy or an imprinting center defect.
Deletions: Deletions causing PWS are categorized as either large, small or microdeletions. Large deletions are often between 4 to 6 Mb and occur in one of two regions. Deletions extending from the D15S541 region to the D15S12 region are often considered class I, while those ranging from D15S543 to D15S12 are categorized as class II. These are collectively the most common type of deletion observed in PWS, and both ultimately inhibit the functions of the imprinting center. Small deletions occur within a smaller range, and microdeletions often occur in areas such as an SNRPN gene exon 1 deletion, which is also known as an imprinting center mutation or defect.
Imprinting center defect: ICD is a subset of deletions most commonly known for deletion of imprinting region exon 1. At the moment, the only available method of detection for an ICD is through DNA testing.
Uniparental disomy: UPD is primarily characterized by a lack of paternal input. In one of the most common forms of the condition, chromosome 15 displays only maternal copies. This condition is often referred to as UPDmat and occurs through a mechanism known as trisomy rescue, in which a combination of a meiotic and somatic abnormal events results in either a heterodisomy or an isodisomy.
Angelman syndrome
Angelman syndrome is often caused by issues with the ubiquitin ligase UBE3A gene located on chromosome 15q. This condition commonly occurs due to activation of only the maternal copy of the gene, with either a missing or defective paternal gene; however, it can also be caused by the inheritance of two paternal genes.
Symptoms of AS can first appear around six to 12 months of age and can include difficulty walking, lack of speech and seizures, among others. There is currently no known cure for the condition.
Current research focuses on the development of mouse models as a way to better understand the development and progression of AS. Humans and mice have similar UBE3A loci, which makes the mouse a viable preclinical model preceding the production of potential translational therapeutics.
Dup15q syndrome
Dup15q Syndrome, also referred to as maternal 15q duplication syndrome, is a condition caused by the presence of at least one extra chromosome 15 region (15q11.2-q13.1). Dup15q typically only occurs when the duplicate copy is maternally inherited.
Some common characteristics of Dup15q syndrome include hypotonia, intellectual disabilities, autism spectrum disorder and epilepsy. Currently, treatment options for the condition are limited to treatment of specific symptoms and surveillance, as well as genetic and prenatal testing to monitor development throughout pregnancy.
There are two forms of Dup15q syndrome that are commonly recognized:
Maternal isodicentric chromosome 15: In this form of Dup15q syndrome, two extra maternal copies of 15q11.2-q13.1 are present, which results in tetrasomy. This form of the condition accounts for approximately 60% to 80% of Dup15q syndrome diagnoses.
Maternal interstitial duplication: This subset of the condition is characterized by the presence of one additional copy of 15q11.2-q13.1, resulting in trisomy for this region of chromosome 15. Maternal interstitial duplication is less common than isodicentric chromosome 15 and accounts for only about 20% to 40% of diagnoses.
The 15q chromosome contains several regions known as segmental duplications, which have a higher susceptibility to rearrangement and thus mutation. Several genes of interest that may contribute to disease progression have been observed in this region, including UBE3A, GABRB3, GABRA5, GABRG3, and HERC2.
UBE3A: This is the same gene that is affected in Angelman syndrome. However, in Dup15q syndrome, it is suspected to play a role in symptoms including intellectual disability, anxiety and a reduced threshold for seizures, which is likely due to an increase in neuronal expression of the gene.
GABRB3, GABRA5 and GABRG3: These genes encode for receptor subunits of the GABAa receptor, a ligand-gated ion channel that plays a major role in synaptic transmission throughout the central nervous system. It has been suggested that this gene may therefore play a role in seizures during Dup15q symptom presentation.
HERC2: This gene is an E3 ubiquitin ligase, part of a subset of genes responsible for interaction with E2 ubiquitin enzymes and a corresponding target protein in order to accomplish transfer of ubiquitin from E2 to the protein. Current literature suggests that pathogenic variants of this gene may lead to neurodevelopmental complications, which is supported by the increased neuronal expression of HERC2 in individuals with Dup15q syndrome.
Therapeutics
Treatment of PWS often includes approaches such as establishing nutritional supplementation for infants, human growth hormone treatment, sex hormone treatment, implementation of behavioral therapies and provision of mental health resources, among other possibilities. While there is not one streamlined approach for treating PWS, there is a repertoire of available resources that aid in early detection and improved quality of life.
AS does not have one specific treatment method. Therapeutic approaches focus primarily on addressing specific side effects of the condition, including seizures, anxiety and gastrointestinal complications, among others. This typically involves implementation of behavioral, dietary, physical or occupational therapies, as well as consultation with physicians to identify additional effective symptom management options.
Similar to both PWS and AS, Dup15q therapeutic approaches are highly diversified depending on clinical manifestation and disease progression. Employment of advice and care from clinical professionals is highly emphasized, and resulting care is individualized depending on the specific needs of each patient.
Resources
While there is still much to be discovered about 15q conditions, there are various resources available for those experiencing these conditions, as well as those who simply want to gain a deeper understanding of current clinical progressions toward treatment of PWS, AS and Dup15q syndrome.
-
This page on the Eunice Kennedy Shriver National Institute for Child Health and Human Development has resources for patients, researchers and physicians about PWS. It includes information about patient advocacy groups, foundations that support elimination of PWS, and organizations that offer support to patients and family members experiencing PWS diagnosis.
-
The Angelman Syndrome Foundation offers information about behavioral resources, counseling and familial support. It also has a podcast, which provides education about the condition.
-
The Dup15q Alliance is an organization that provides information about current research, clinical trials and even volunteer opportunities to foster a greater, more widespread understanding of Dup15q syndrome.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.

Flipping lipids and slime molds
A dull first job nearly pushed JBC associate editor Todd Graham out of science. Then a slime mold project changed his path. Now, he studies membrane biology and reflects on discovery, persistence and mentoring through uncertainty.

How smelling death alters worm behavior
Researchers have found that the roundworm C. elegans can smell death, and it changes how the worms behave, reproduce and age.

A chance encounter with the lab
Payton Stevens never planned to become a pancreatic cancer researcher. A temporary job set him on a path from rural Kentucky to leading research on Wnt signaling and metastasis, where he now pairs discovery with mentorship and science advocacy.

Light-activated small molecule could transform eye infection treatment
Contact lenses raise the risk of infectious keratitis, a leading cause of blindness worldwide. A biotech company is commercializing a light-activated therapy using a ROS-generating molecule to rapidly kill microbes in the cornea to preserve vision.
The molecular orchestra of memory
Calcium, calmodulin and calcium/calmodulin-dependent kinase II form a molecular axis that turns fleeting neural activity into lasting memories. New research shows how memories are stabilized, and possibly even protected or repaired.