From genes to hope

Niemann‒Pick disease is an umbrella term for a rare, heterogeneous group of inherited metabolic disorders characterized by the harmful accumulation of lipids, particularly cholesterol and sphingolipids, within cells of the brain, spleen, liver, lungs and bone marrow due to defective lysosomal function. Hence, the disease is also part of the family of lysosomal storage diseases, which are characterized by defective lysosomal function leading to accumulation of various substrates in excess in cells.
Excess lipid accumulation in NPD leads to cell death and, thereby, the malfunction of affected organs. People with NPD experience symptoms related to progressive loss of function of nerves, the brain and other organs.
Today is Global Niemann‒Pick Disease Awareness Day. It’s an observance dedicated to bringing the world’s attention to the complex condition to accelerate research and ultimately the delivery of therapies and cures.
This article explores the first identification of the disease, its three main types, its molecular mechanisms and recent research published in the American Society for Biochemistry and Molecular Biology journals with promising strides toward effective treatments.
Namesakes
The disease’s name is an eponym coined in the late 1920s to recognize the pioneering work of German pediatrician Albert Niemann and German pathologist Ludwig Pick to understand the disease.
In 1914, Niemann described the first NPD patient, an Ashkenazi Jewish infant who presented with massive enlargement of both the liver and the spleen and rapidly progressing neurodegeneration that led to her death at 18 months of age. Niemann, who is often confused with a chemist of the same name who isolated cocaine, died at the age of 41 in 1921.
Pick’s post-mortem tissue examinations and histology preparations revealed that the disease that later would bear his name was distinct from the previously described Gaucher disease in 1926. Pick was imprisoned by the Nazis during World War II and died in a concentration camp in 1944.
Multiple forms
NPD occurs with an incidence rate of less than one in 100,000. It can manifest at any age but mainly affects children. The disease currently has no known cure and treatment is focused on helping people live with their symptoms.
The disease is categorized into several types, with Types A and B being the most common, followed by Type C, each with its unique characteristics and symptoms.
-
Type A is a severe form that typically manifests in infants, causing severe neurological, and visceral problems. Unfortunately, these children seldom survive past 2 to 3 years of age.
-
Type B tends to present later in life and primarily affects the liver and spleen. Individuals with Type B often have a longer life expectancy compared to Type A.
-
Type C is the most complex form and can manifest at any age. It affects multiple organ systems, including the brain. Its symptoms vary from mild to severe, posing significant diagnostic and management challenges.
Molecular mechanisms
The underlying reason for NPD depends on the type, although all are due to single gene mutations inherited in an autosomal recessive pattern.
Type A and B are caused by missense and nonsense mutations in the sphingomyelin phosphodiesterase-1 gene, or SMPD1, located within the chromosomal region 11p15.4.
The SMPD1 gene is preferentially expressed from the maternal chromosome and encodes the enzyme acid sphingomyelinase, or ASM. ASM has its highest activity at acidic pHs and catalyzes the hydrolytic cleavage of sphingomyelin in lysosomes to produce phosphocholine and ceramide. Sphingomyelin is a major component of cell membranes and a principal phospholipid of the myelin sheath.
Within lysosomes, ASM interacts with other lipid hydrolases and performs an essential housekeeping function by maintaining proper sphingolipid homeostasis and participating in membrane turnover, a critical mechanism that maintains cell surface area. When cells are subjected to stress, ASM rapidly translocates from lysosomes to the outer leaflet of the plasma membrane and hydrolyzes sphingomyelin into ceramide. This causes the reorganization of membrane lipid microdomains, or raft structures, and stimulates downstream signaling events including the ceramide-mediated signaling pathway.
Mutations in SMPD1 responsible for NPD A and B cause the ASM that is expressed to be functionally deficient. NPD A results from defective ASM with minimal catalytic activity. Therefore, people with Type A experience rapid neurologic degeneration from the accumulation of sphingomyelin in ganglion cells.
Type B patients do not experience this symptom because the mutated gene still encodes for a defective version of ASM with considerable catalytic activity. They experience less severe sphingomyelin accumulations in the spleen, liver and lungs.
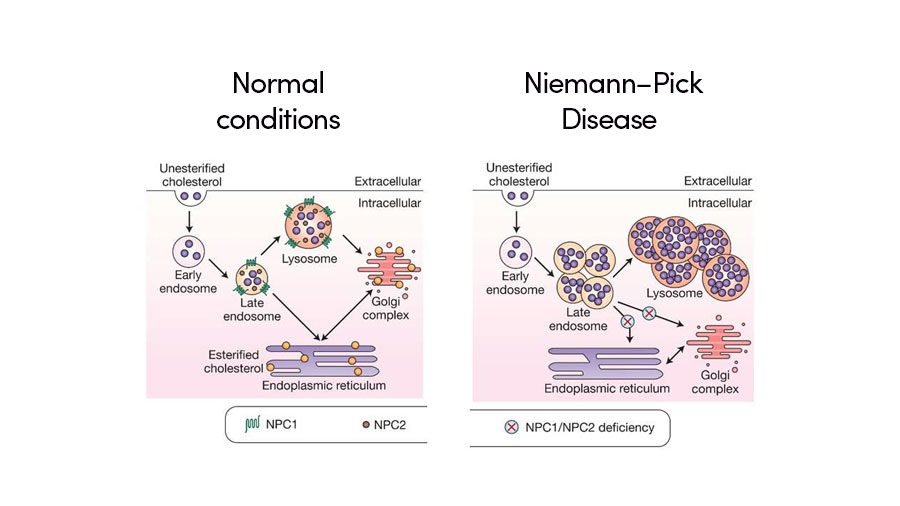
NPD Type C is caused by the genetic mutation on either genes NPC1 or NPC2. In fact, 95% of patients have mutations in the NPC1 gene. These genes are located on chromosome 18.
NPC1 encodes a late endosome-resident protein that transiently associates with lysosomes and the trans-golgi network. NPC2 encodes a small soluble lysosomal protein that binds cholesterol with high affinity.
Both of these proteins are responsible for cholesterol transportation in late endosomes, a major sorting compartment of the endomembrane system within eukaryotic cells, and lysosomes. With a disruption in either of these genes, the late endosome and lysosome of affected cells begin to accumulate unesterified cholesterol and lipids. Excessive accumulation of cholesterol in both the late endosome and lysosome results in irregular intracellular transport, cell signaling and nerve conduction.
Recent breakthroughs
Researchers have made significant progress in recent years in understanding the disease mechanisms and developing targeted treatments.
Notable breakthroughs include:
Improved understanding of pathophysiology: Recently, Marko Kosicek and a group of researchers from Croatia were able to show for the first time that the N-glycome of the lysosomal glycocalyx of NPD Type C disease model cells is considerably different from unaffected cells. This indicates changes in the lysosomal glycocalyx may be a useful avenue for restoring lysosomal activities in dysfunctional lysosomes in patients. Read the full study published in the journal Molecular & Cellular Proteomics
Enhanced diagnostic tools: Precise diagnostic tools, such as biochemical testing and genetic sequencing, have emerged, enabling rapid, accurate and earlier detection of NPD.
Na Lin and a group of researchers from China recently developed a method that analyzes the plasma 7-ketocholesterol by LC-MS/MS to diagnose NPD. This is a simple, quantitative and highly sensitive method for the detection of all types of NPD. Read the full study published in the Journal of Lipid Research.
Masamitsu Maekawa and a group of researchers from Japan recently developed a rapid, convenient and non-invasive chemical diagnosis method for NPD using LC/MS/MS. In their project, they simultaneously analyzed five conjugated cholesterol metabolites in the urinary that are biomarkers for NPD. Read the full study published in the Journal of Lipid Research
Therapeutic advances: While there is currently no cure, several promising treatment options are under investigation. Researchers are actively exploring various approaches — including enzyme-replacement therapy, substrate-reduction therapy and gene therapy — to address the root causes of NPD and alleviate its symptoms.
Enzyme-replacement therapy involves replacing the missing or deficient enzyme and has shown potential to treat Type B. Recombinant human ASM expressed from Chinese hamster ovary, or CHO, cells are currently in the phase 1/2 pediatric trial and phase 2/3 trial for adults. Read more about this.
Substrate-reduction therapy aims to reduce the production of harmful lipids in the body, offering promise for Type C. Kanagaraj Subramanian and a group from the U.S. demonstrated that the histone deacetylase inhibitor valproic acid corrects the folding and trafficking defect associated with I1061T-NPC1, leading to restoration of cholesterol homeostasis. Read the full study in the Journal of Biological Chemistry
Concluding thoughts
NPD is a formidable challenge, but recent research is unraveling its complexities and opening doors to innovative treatments. By understanding the molecular mechanisms, exploring therapies like enzyme-replacement therapy, and harnessing the power of gene editing with CRISPR‒Cas9, we pave the way for a future where NPD is no longer an insurmountable obstacle but a condition with hope on the horizon.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.