
The scramble for protein nanopore sequencing

When Jeffrey Nivala was a graduate student, his work station was a vibration-dampening table covered in electronic instruments: a patch clamp amplifier, an analog-to-digital converter, a perfusion system. All those trappings were trained on one tiny focal point: a tube roughly a nanometer in diameter and the minuscule trickle of electricity that flowed through it.
“I spent most of my Ph.D. chained to one of these things and trying to squeeze data out of it,” Nivala said.
The tube, called a nanopore, was formed by a ring of proteins tunneling through a lipid membrane. After setting up an experiment, Nivala would sit and wait, hoping for a group of seven proteins to self-assemble and poke through the lipid membrane, opening a route for ions to follow. When ions, or charged particles, move, they create an electrical current. After his equipment detected a current, Nivala would watch it intently, waiting for minute fluctuations that would indicate that a macromolecule was passing through the pore.
Nowadays, a pocket-sized device that plugs into a laptop’s USB drive can deliver comparable information — from hundreds of pores at once. Nivala said, “A student in my lab now can collect more data in an afternoon than I ever did in my Ph.D.”
After decades as an aspirational concept, nanopore DNA sequencing has been fully realized. Commercially available nanopore flow cells have been used to sequence DNA by astronauts on the International Space Station, biologists skiing across Iceland and geneticists piecing together the last missing fragments of the human genome.
Now, the biophysicists and engineers who developed the DNA sequencing technology are turning their attention to proteins, which are substantially more difficult to analyze with a nanopore. Researchers around the world are working to surmount the problems with proteins, hoping to make single-molecule proteomics a reality.
How DNA nanopore sequencing works
The engine that powers nanopore sequencing is a pair of liquid compartments separated by a membrane, with a positive charge on one side and a negative charge on the other. When a protein nanopore inserts into the membrane, it opens a path for ions to flow from one side to the other, responding to the difference in charge between them. (This difference in charge is measured in voltage; the rate of ion flow in response to a voltage difference is measured in current.)

device from Oxford Nanopore, are small and highly portable.
Most of the ions that make up the current are charged single atoms such as chloride and potassium. When the pore is open, ions make the crossing easily, slipping through in nanoseconds. Larger molecules also can feel an electrical pull. The negative charge of DNA’s phosphate backbone will draw it toward a positive charge, and therefore into a pore, just as the difference in charge pulls the chloride ions. But once that strand of DNA enters the pore, it takes up most of the space inside, allowing relatively few smaller ions to squeeze past and cross the membrane. That reduction in the movement of charged particles is reflected in a change in current picked up by electrodes on opposite sides of the membrane.
Each of the four DNA bases has a different chemical identity and blocks ions to a different degree. Like four different people casting distinct shadows as they pass through a door from a bright room into a dark one, the nucleotides can be identified by their signature current changes.
Left to its own devices, a linear strand of DNA would zip through the pore rapidly, although not quite as fast as the ions around it. Aleksei Aksimentiev, a biophysicst at the University of Illinois at Urbana–Champaign, said that slowing DNA down was key to the development of nanopore sequencing. “If it goes too fast, you just don’t have enough ions … so you might make a huge error when you determine the average value of the current.”
To measure the change in current with more confidence, researchers borrowed motors such as polymerases and helicases from cells’ DNA replication machinery; these enzymes bind to the DNA and advance just one nucleotide at a time, forcing it to take measured, slow steps.
In addition to finding ways to make each fluctuation in current last a little longer, researchers also have developed algorithms to interpret the changing current and determine in real time which nucleotides are passing through the pore. The technology can collect long sequence reads from a single molecule of DNA, letting researchers draw conclusions that other technologies don’t offer.
Aksimentiev is one of the many researchers who hope to apply the same principles to achieve protein sequencing. His lab has been working on the problem since transitioning from DNA sequencing in 2016. If they succeed, the work might lead to a portable device that could be used to determine the amino acid sequences and post-translational modification patterns of much longer pieces of single proteins than currently are possible using mass spectrometry, which would allow researchers to study signaling and isoform expression in greater detail than ever.
But it won’t be easy, Aksimentiev said.
Biochemical challenges
“There are lots of challenges with proteins,” Aksimentiev said. “In particular, they’re not uniformly charged; they’re not linear, most of the time they’re folded; and there are 20 amino acids, plus a zoo of post-translational modifications.”
While the backbone of DNA carries a negative charge, a polypeptide backbone is neutrally charged at physiological pH, and its amino acid side chains come in a rainbow of sizes and polarities. Because of this heterogeneity, whereas DNA reliably carries a negative charge, many proteins do not.
All that heterogeneity doesn’t just make it difficult to move a protein through the pore; it also may make for a more complicated analysis. Each of the four DNA bases produces a signature change in current, and it is much easier to differentiate among four signals than 20 — or, counting the possibility of post-translational modifications, many more.
Researchers have begun to show that at least a few distinct amino acid signatures exist, but they have not yet resolved all 20. In the journal Nature Biotechnology in early 2020, researchers in Abdelghani Oukhaled’s lab in Cergy, France, collaborating with Aksimentiev’s lab, reported that they could differentiate between the nanopore current signals from short peptides that varied by just a single amino acid. Each peptide was eight amino acids long, with seven identical negatively charged residues and one that varied. The team found a predictable interaction between the size and charge of that single variable residue and the way the current changed as the peptides passed through the pore.
When they tried to measure a mixture of all 20 peptides, the researchers found that they could pick out a few of the peptides with high confidence. According to some of their calculations, coaxing the peptides to linger longer in the pore would enable them to identify even more with confidence.
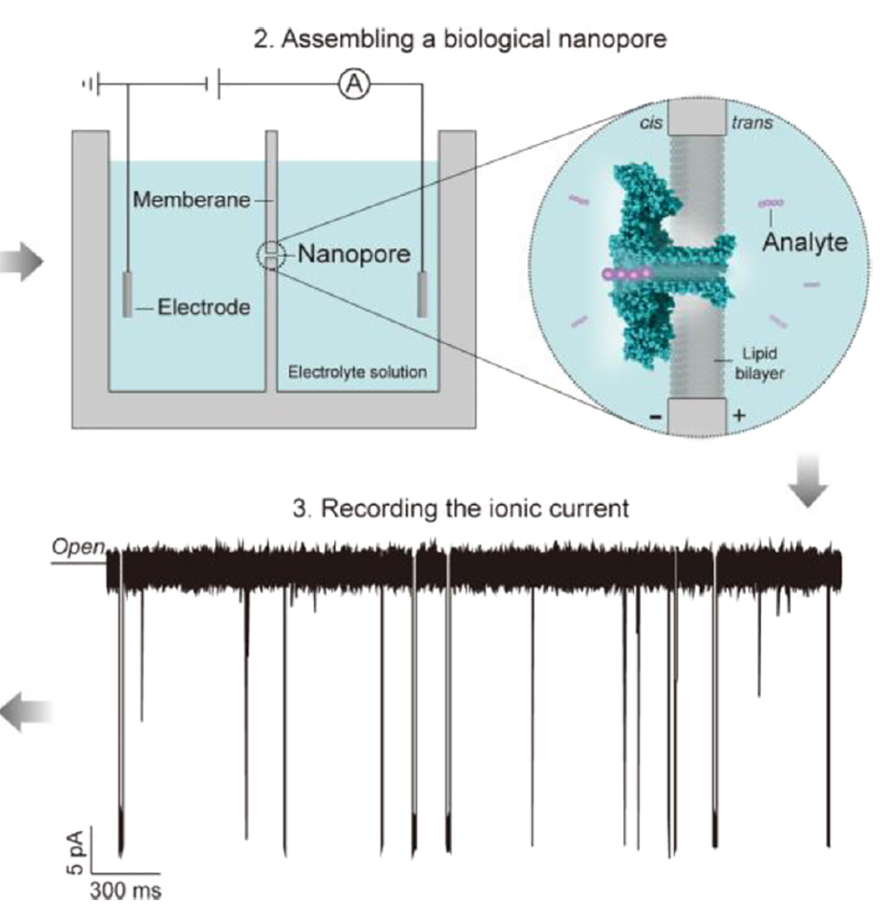
To make larger proteins pass through the pore in an orderly way, researchers must make them more linear, smoothing away secondary and tertiary structures, without damaging the structure of the proteins that make up the pore or the integrity of the lipid bilayer it is embedded in. At this point, Aksimentiev said, enough solutions are being proposed, tested and optimized that he thinks “unfolding the protein and feeding it through is probably not going to be the critical part” of achieving protein sequencing.
Unfolding the protein, feeding it through
As a grad student at the vibration table, Nivala, who is now a research assistant professor at the University of Washington, focused on the problem of turning a folded protein into a linear molecule that fits neatly into a pore.
“My Ph.D. was focused on showing the first proof of principle of using these motors to analyze these full-length, intact protein strands with a nanopore,” Nivala said. “At that time, the writing was on the wall that the big challenges in nanopore-based DNA sequencing had been or were about to be solved.”
Working in the lab of University of California, Santa Cruz, biophysicist Mark Akeson, Nivala developed a way to use unfoldases — enzymes that, in their typical context in the mitochondria, unfold proteins in preparation for proteolysis — to pull proteins through an alpha-hemolysin nanopore.

of knitting, the force breaks up weaker interactions that hold the protein
in its structure, rendering the protein linear.
The unfoldase he used, ClpX, does to a structured protein what a determined tug on a stray thread does to a sweater; it applies force to a loose end that overcomes the stabilizing power of internal interactions within the protein.
Nivala developed a C terminal protein tag that combined a highly charged stretch of amino acids and a protein motif recognized by ClpX. Because of the charged residues, a voltage difference would attract the end of the protein through the pore just far enough to encounter ClpX stationed on the opposite side. Then, Nivala said, the enzyme “would grab on to the tail and start pulling.”
The motor protein would do the rest, tugging along the peptide backbone with enough force to unravel the structure on the other side of the membrane and pull the protein through. While this solved the problem of moving a noncharged protein through the pore, it also illuminated some challenges. For one thing, the pore that Nivala used was long and narrow; the portion of its tube that was the tightest squeeze, where current was determined, had space for as many as 30 amino acids at a time. Each of them could contribute to the overall flow of ions. Instead of trying to identify a single silhouette against a lighted door, imagine working with a shadow formed by a line of dozens of figures. Nivala said, “It ends up being a very intractable problem to deconvolve that into amino acid sequences.”
Scientists had solved a similar challenge in DNA sequencing, but it was less complex because nucleotides are larger in size and smaller in number. They also had assistance from the stepwise work of the enzyme motors, which changed the signal by exactly one base every time. Although ClpX pulled its target proteins in regular steps of about a nanometer, one nanometer of protein backbone can include anywhere from five to eight amino acids. Nivala detected changes in current that showed some features of the test protein he was using, but he was unable to translate those changes back into a sequence of amino acids.
Cees Dekker, a physicist at Delft University of Technology, called the work “a very elegant idea … that faces some technical challenges.”
In the years since, researchers have experimented with many other approaches to linearize proteins. Nivala’s lab is examining unfoldases that might meet sequencing needs better. In Nature Chemistry, in November 2021, Giovanni Maglia’s lab at the University of Groningen reported that they had engineered a 42-protein complex that resembled an inverted soda bottle with its neck forming the pore in the membrane. The molecular machine, which combines a proteasome with a nanopore, uses motors from the proteasome component to drive an analyte protein into the pore.
Some researchers are working on alternative motors, enzymes that act on DNA instead of proteins. Dekker’s lab has worked for decades on what he calls “creative ways to get sequence information from DNA.” Perhaps that familiarity is what gave them the idea to use DNA to get sequence information from proteins. Late in 2021 in the journal Science, the team reported an approach that used a DNA helicase to draw a chunk of protein through a nanopore complex called MspA, which previously had been used for DNA sequencing.
Conjugating a strand of DNA to a negatively charged short peptide, they applied voltage to make the conjugated molecule translocate through the pore. Then they added DNA helicase that would pull in the opposite direction, dragging the protein–DNA conjugate backward. The helicase’s steps in one direction and the pull of electricity in the other added up to a seesaw that put tension on the linear molecule, holding it in the pore as ions whizzed by. That gave the electrode measuring current enough time to get a reliable reading at each step.
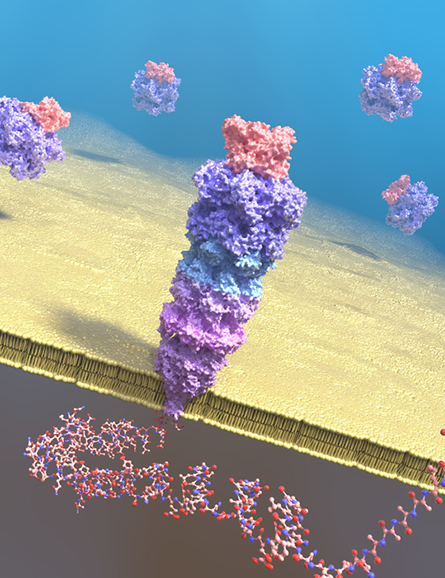
“What we have established is an identification tool that can identify certain proteins with great confidence; even single amino acid mutations can be very well resolved,” Dekker said. “What we have not achieved yet is a de novo sequencing tool.”
The researchers were able to analyze a small chunk of protein topping out around 25 amino acids. The peptide used in these first experiments was negatively charged, and Dekker said that the DNA–peptide conjugate molecules are costly to synthesize for now.
Despite these challenges, Yi-Tao Long, a professor at Nanjing University who has worked in nanopore peptide analysis since the early 2000s, said the work has attracted significant interest to the field. “It is a milestone towards nanopore technology to directly sequence proteins.”
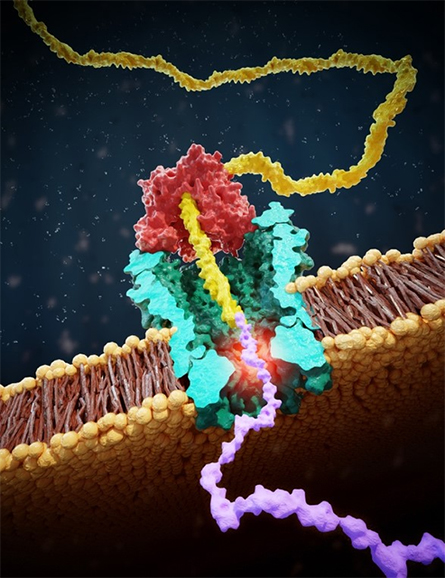
in yellow, protein in purple.
Interactions with the pore
The Dekker lab’s work also illuminated the importance of molecular modeling to understand what really happens inside a pore during a protein sequencing run. While testing whether the DNA conjugation system could help recognize a peptide that varied by just a single amino acid, Dekker’s team observed that the peptide with the largest variable amino acid they tried, tryptophan, caused a peculiar signal.
“If you block part of the ion flow, you get a lower current. That’s the intuition we built from,” Dekker explained. But in the case of tryptophan, “You would expect, well, its volume goes up and (the current) goes down. But this saw more of a wiggling signal.”
Tryptophan is bulky; it makes sense that wedging it into a small pore would block most ions from going past. The team expected that it would produce a drop in current, which they thought would recover to the baseline. The current they observed instead dropped dramatically but then went up, indicating that the pore was becoming more open.
Dekker approached Aksimentiev, a longtime collaborator, to help make sense of the strange readout.
When the structure of a pore is known at sufficient resolution, researchers can use supercomputers to run complex, high-precision simulations of its behavior. Aksimentiev’s lab specializes in this work. “Every atom of the protein, every atom of every water molecule, every ion, peptide: everything is there,” Aksimentiev said. “And once we have this truly atomic-scale representation of the experimental system … just like an experiment, we can see ions moving through the pore and measure the current.”
In contrast to scientists in the wet lab, though, the team can peer into the pore moment by moment to find out why the current is changing as it is. Aksimentiev’s group found that tryptophan formed a hydrophobic interaction with the amino acids in the inner wall of the pore.
“The intuitive argument that if you have something bigger in the pore, that blocks the current more, is correct,” he said. “It’s just that once the tryptophan has passed … it sticks to the wall and brings everything to the side, actually making more space for the ions to pass.”
This peculiarity gave tryptophan a highly recognizable signature, he added. “One can see how one can potentially engineer a pore to have those specific interactions and look for difficult-to-distinguish stretches.”
Would this phenomenon happen when any peptide containing tryptophan crossed through this pore? Aksimentiev thinks it will depend on the peptide sequence and local chemical environment. In any case, the finding illuminates the complexity of factors affecting the current readout.
Long said that researchers are accustomed to thinking about current as only a function of volume: In other words, an amino acid’s size is the most important determinant of how ions flow past. But, he said, that’s probably an oversimplification. Every individual amino acid has the potential to interact with the pore, and so does every ion. These interactions may carry information, but also may increase the complexity of protein-sequencing currents dramatically. And what’s more, there are many different pores to choose from.
Many pores
“A polymer is like the notes of music,” Long said. “The nanopore is like a musical instrument, a keyboard.”
As the polymer passes through the pore, the current is akin to a tone. The same analyte might evoke different currents from different pores, just as a melody sounds different when played on an accordion or a music box.
When, as a postdoc in 2002, Long began to analyze protein structures using a nanopore, his team initially used alpha-hemolysin, which at the time was the only pore in use for DNA sequencing. As it happened, the lab next door at the University of Saskatchewan had a convenient plasmid on hand; the PI had trained in the lab that discovered a bacterial channel toxin called aerolysin and shared it with them. Long’s lab uses it to this day.
Hemolysin and aerolysin are just two examples of a vast protein family (see sidebar: “Many pores, many structures”). Multiple labs have published on pores borrowed from various other bacteria: hemolysin, aerolysin, fragaeceatoxin and more. These proteins have diverse characteristics. Some are barely wide enough to squeeze a polymer through the central pore, while others are broad tubes. Others are shaped like funnels that narrow to one critical sensing region.
According to Long, the complexity of how pores interact with proteins once inspired researchers in the field to try using solid-state nanopores built from carbon nanotubes or other fairly homogeneous nonbiological materials. These nanostructures have advantages; they hold up in heat, pH and detergent conditions that can linearize a protein but would destroy a biological nanopore and its lipid environment.
In addition, researchers hoped that engineered pores might yield a smoother interior surface, minimizing the potential for interactions with biomolecules. However, Long said, “That’s not the case. Nobody can make an atomic structure smooth.”
The idiosyncrasies of proteins, he added, can give designers more room to experiment. “With 20 amino acids to play with, we can make different structures we want.”
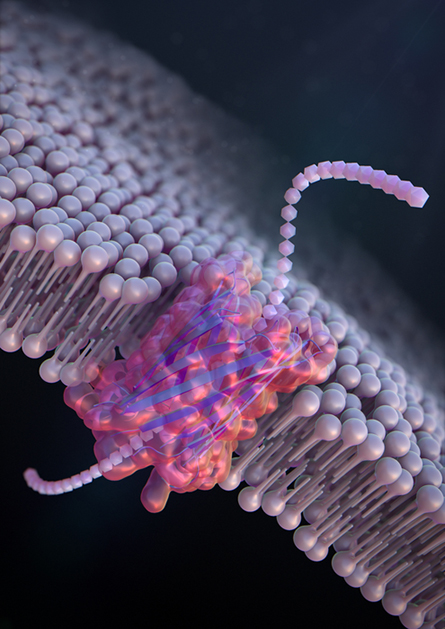
reported in Nature Nanotechnology last year.
“Nature has lots of different pores that it’s using for lots of different reasons,” Nivala said. “We’ve probably barely tapped into what nature’s already built.” Meanwhile, improved tools for protein design have led to researchers “starting to engineer protein nanopores from scratch that you can custom-tailor for different applications.”
A spokesperson for the company Oxford Nanopore said that when working with a new bacterial pore, the company makes and tests thousands of mutants, aiming to optimize protein expression, interaction with DNA and signal clarity during sequencing.
Although Oxford Nanopore, a juggernaut in the DNA sequencing field, declined to comment on protein sequencing for this article, Nivala said that his lab works closely with the company. He said the pore currently used in Oxford Nanopore’s DNA-sequencing flow cells, a secretion channel called CsgG, may not be perfect for protein sequencing. But whatever drawbacks it may have are outweighed, for now, by its ease of use.
“If we are able to do anything useful, some other lab could pretty much instantly then adopt that technology and that technique,” he said. “They don’t need to be nanopore experts.”
Additionally, because Oxford’s technology is mature, Nivala’s team can focus easily on collecting and analyzing data, not on preparing nanopore wells. That’s a good thing, he said. “We’re going to need lots and lots of data … to develop machine learning algorithms that can make sense of these signals and decode them back into amino acid sequences.”
Problems to solve
Researchers have developed software tools that reliably can identify a short test peptide from a list of options. They still have a long way to go to achieve true protein sequencing. Software that can decrypt the tiny fluctuations in current, just a millionth of an amp at a time, will be as important as choosing the proper molecular machinery.
The race is on to develop algorithms that can determine a sequence starting with no information about it. If that goal can be realized, the combination of single-molecule resolution with long reads could open a door to single-molecule studies of proteins that are currently impossible. Researchers are interested in applications such as antibody sequencing and investigating how multiple post-translational modifications on a single protein, such as a histone, could impact its function.
Instead of conclusively identifying every single amino acid, some researchers have proposed protein fingerprinting, which would pick out individual easy-to-identify amino acids that have strong signatures — such as the wiggle that Dekker’s lab observed with tryptophan — and use them as landmarks to infer a protein’s identity. Sample preparation that would tag individual amino acids specifically with distinctive chemical labels could help with an approach like this.
Even with their existing shortcomings, researchers say, nanopore protein sequencing systems can already be used for interesting applications such as scanning short peptides for negatively charged post-translational modifications.
And researchers are confident that more advances are just over the horizon.
“It’s all coming together,” Aksimentiev said. “It’s a very exciting time for single-molecule protein sequencing.”
Many pores, many structures
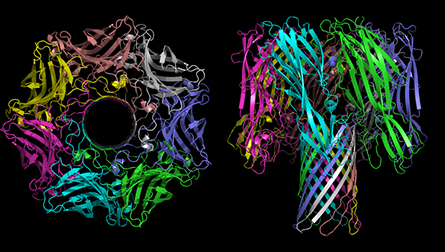
The sequencing community regards pore-forming proteins as a useful biochemical tool. Another research community, one with few overlaps, studies why bacteria make the proteins, their structure and how they work.
Melanie Ohi, a structural biologist at the University of Michigan who studies the assembly of numerous protein complexes, keeps several boldly colored images of pore complexes tacked to the white board in her office. The largest depicts VacA, a pore-forming protein from the stomach pathogen Helicobacter pylori, whose structure her lab solved a few years ago. She said, “These toxins … are beautiful. They are the prettiest molecules you’ll ever look at.”
Beyond their pleasing aesthetics, Ohi is interested in what she called toxins’ “fascinating ability to live in two very different environments.”
Many bacteria, including H. pylori, secrete the subunits that make up protein pores into their environment, meaning that the toxins must be water soluble. But to function, those soluble subunits also must find one another, assemble into that pleasing symmetrical ring, and accomplish a biophysically tricky insertion into a membrane, requiring that they show a hydrophobic face. The cryo-EM revolution, Ohi said, is helping researchers to understand the many conformations these proteins can adopt at progressive stages of this attack.
Depending on its size and selectivity, the pore that eventually forms can render a membrane permeable to ions or to larger molecules. “A lot of bacteria have these types of toxins,” she said. “They’re used either to create a niche for the bacteria to live and grow in; or as a way for bacteria to deliver effectors into a host cell; or as bacterial warfare.”
Pores beyond protein sequencing
Yi-Tao Long, a professor at Nanjing University who has been working on developing protein analysis methods that use nanopores since the early 2000s, said that he does not expect pores to overtake mass spectrometry for proteomics completely.
There are uses for which mass spec has downsides, such as differentiating amino acids or modifications with identical mass, where nanopores shine. They also work fast; a researcher with a nanopore can detect biological systems in real time, “like a movie,” Long said. Still, “Mass spec is a fundamental technique for protein sequences. Nanopore sequencing cannot be a replacement. It can be complementary, especially for some application environments.”
In the world of DNA sequencing, by comparison, both the single-molecule long reads obtained through nanopore sequencing and the massively parallel short reads obtained through next-generation sequencing have advantages and are useful for different types of study.
Long said some of the most exciting applications for nanopores open completely new capabilities. For example, he is interested in using nanopores to make enzymatic synthesis faster at an industrial scale. Whereas chemical syntheses can use heat and stirring to increase the frequency with which reactants collide, speeding reactions, enzyme-catalyzed systems can’t stand up to heat and vigorous stirring. Instead, Long said, what if researchers found ways to embed catalytic sites into the walls of a pore and then draw reactants in?
He also emphasized that while sensing specific amino acids is useful, nanopores also can be used to sense other types of analytes, such as environmental contaminants.
Structural biologist Melanie Ohi said that many basic scientists who study bacterial pores are interested in the possibility of using them for drug delivery. “A lot of these toxins are very cell-specific,” she explained. Although it will take more research to determine whether their specificity comes from being released near those cells or from recognition of cell surface molecules, “It seems like it could be very powerful if you could take the bacterial toxin that is delivered to a very specific cell and re-engineer it to deliver a drug.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.