Slipping past the proofreader

In the early 2000s, when experts believed human coronavirus infection caused nothing worse than the common cold, Mark Denison struggled to land adequate federal grants to support his lab at the Vanderbilt University Medical Center.
A virologist and clinician, Denison had been studying coronaviruses since 1984, when only two of the seven coronaviruses currently known to cause disease in humans were known. Those two, along with several other coronaviruses, cause colds.

Denison initially found the virus he studied, which affects only mice, interesting because it leads to a mouse version of multiple sclerosis. Along the way, he became interested in how the virus replicated — but persuading funders to support his work was a real challenge.
In early 2003, he and his wife, Laura, were on vacation in Florida having a difficult conversation. “I think the work is important. I think the models are important,” he recalled saying. “(But) I don’t know if I can maintain a career.”
That was the day he learned about the pathogen behind the deadly respiratory disease SARS, which had been making headlines.
“I’m literally on the beach with my wife, and someone came down from the hotel and told me that I had a phone call,” he said. The call was from a colleague, sharing the news that the pathogen responsible for the disease had been identified, and it was a coronavirus.
Severe acute respiratory syndrome swept from southern China into 26 countries in 2002 and 2003, killing about 10% of the roughly 8,000 people it infected. Although the outbreak was contained, coronaviruses were recognized suddenly as a serious problem of potentially pandemic proportion.
Almost two decades later, the work done in Denison’s lab has turned out to be instrumental in developing a molecule, called remdesivir, that entered large-scale clinical trials just weeks after SARS-CoV-2, the coronavirus that causes COVID-19, was identified.
The molecule, it seems, can overcome coronaviruses’ superpower: their genome-proofreading ability. This ability isn’t found in other RNA viruses, and it makes coronaviruses resistant to a majority of drugs used against other RNA viruses.
Researchers expect to learn in the next few weeks whether remdesivir is an effective treatment for patients with COVID-19. In the meantime, Denison’s lab has continued to work to identify other molecules that might bypass viral resistance.
“This is a really key question for us: Is this a new class of drug that can allow us to better design more drugs that can bypass the proofreading function and inhibit the virus?” said Denison, who now directs the pediatric infectious diseases division at Vanderbilt.
Not your average RNA viruses
Like many viruses that cause human disease, coronaviruses have a genome composed of RNA.
When the virus behind SARS was identified, one of the many experimental treatments clinicians tried using was a molecule called ribavirin. At the time, ribavirin was the first-line drug for many RNA viruses.
Ribavirin targets a viral protein called the RNA-dependent RNA polymerase, or RdRp, which is responsible for replicating the coronavirus genome.
Craig Cameron is a virologist at the University of North Carolina at Chapel Hill who studies RdRp molecular mechanisms in picornaviruses. “(RdRp) is a very well-validated drug target,” Cameron said. “And it is one of those targets that actually have the potential of having pan-viral antiviral activity.”

Ribavirin belongs to a class of antiviral drugs called nucleotide or nucleoside analogs.
Exactly how ribavirin works seems to vary from one virus to another, making it a good illustration of the many possible modes of action of nucleotide analogs. Some work by blocking the viral polymerase, terminating a growing strand of RNA. Some are mutagens, which slip into the growing viral genome, letting the polymerase continue, but introduce some molecular ambiguity to the next round of replication that causes a cascade of errors in later generations. Some block metabolic enzymes, preventing the synthesis or processing of real ribonucleotides and thereby slowing down replication.
A systematic review of data from 30 clinical trials, conducted after the SARS epidemic, showed no conclusive benefit of ribavirin — or any other treatment that was tested — and some evidence that ribavirin had done patients harm.
Ribavirin was the first example. Since then, most nucleotide analogs tried against coronaviruses that cause SARS and Middle East respiratory syndrome, or MERS, have not been effective treatments. Referring to ribavirin and the classic nucleoside analog 5-fluorouracil, which works as a mutagen, Denison said, “Coronaviruses are completely, entirely resistant to those drugs. You can soak them in it, and they have no effect.”
Vigorous viral proofreading
Bruno Canard is the principal investigator of a viral replication group at the French National Centre for Scientific Research. Before the SARS outbreak, Canard had focused on the structure–activity relationships of nucleotide analogs used to treat HIV and other viruses. Like many virologists, he was inspired by the near miss with SARS to start new research programs.
“We were surprised that ribavirin was pretty toxic and not very effective (against) coronaviruses,” Canard said.

Both the CNRS team in Marseille and Denison’s group in Tennessee set out to understand more about the virus that caused SARS, and one of their major questions was why ribavirin, broadly effective against other RNA viruses, had failed against this one.
The answer lay in the virus family’s large genome and how it evolved to protect itself.
“The reason (RNA viruses) are thought to be so successful is because their polymerases make mistakes,” Denison said. “They lack the ability to correct mistakes, so they generate mutant swarms of viruses that are ready for adaptation in different environments.”
Structural biologists describe RNA-dependent RNA polymerases as resembling a cupped hand, with the fingers and thumb protecting the enzyme’s active site. As each new nucleobase in the template strand enters the active site, the polymerase coordinates a new incoming ribonucleotide by matching it against its counterpart in the existing strand. If the fit is right, the enzyme catalyzes a bond formation in the RNA backbone. If the fit is close enough, many viral RNA polymerases will catalyze a bond anyway. An error rate that can be as high as one mistake per 10,000 bases lets those mutant swarms arise. But for coronaviruses, the replication error rate is lower.
Coronaviruses have some of the longest genomes in the RNA viral world. Whereas their closest cousins have genomes averaging 10 kilobases, coronavirus genomes are three times as long. With so much genetic material to copy, if coronaviruses mutated at the same rate as other RNA viruses, they would accumulate so many mutations that they would barely produce any viable progeny.
As researchers in the field came to understand the coronavirus RdRp better, they found that the polymerase by itself could not explain the disconnect. Virologist François Ferron, a staff scientist at CNRS, has worked on coronavirus replication since the SARS outbreak. “The viral RNA polymerase is quite loose, meaning it has a tendency to make a lot of mistakes,” he said. “Maybe a little bit more than the regular (cellular) RNA polymerase.”

This led researchers to suspect that coronaviruses might have some way of recognizing and correcting errors.
An international team conducting a study of the SARS viral genome identified a number of potential RNA-processing enzymes based on their homology to other known enzymes. In 2006, Canard’s group worked with members of that international bioinformatics team to show that one of those enzymes, like its homologs, could cleave double-stranded RNA and was required for successful viral replication.
“There was a speculation that (coronaviruses) might encode a proofreading function that would allow them to stabilize a big genome, and there was a predicted place in the genome where that might occur,” Denison explained. “That really led us to try the genetic experiments.”
In 2007, his group found that in viruses lacking the protein encoded at that same location, a protein called nsp14, coronaviruses from a mouse model virus accumulated mutations at a rate similar to other RNA viruses. And strains of the virus without nsp14, they found, were sensitive to ribavirin.
Following up on that work, the French group zeroed in on how nsp14 worked in a test tube. “We didn’t work on the virus, like Mark Denison was beautifully doing,” said Canard. “We just concentrated on that enzyme … and we found out that it could actually excise ribavirin.”
The CNRS team published an enzymology study that confirmed that nsp14 can identify and remove mismatches between bases at the end of a growing copy of viral RNA; in 2018 they followed up with confirmation that when ribavirin is incorporated in a growing RNA strand, nsp14 protein can scoop it out of the stalled strand, letting replication resume.
As the researchers worked out the enzymology, a pressing question arose: Did their findings mean that all nucleotide analogs would be useless against coronaviruses?
“This question is actually key right now in the development of nucleoside analog inhibitors,” Canard said.
Anatomy of a molecule: What makes remdesivir unique?
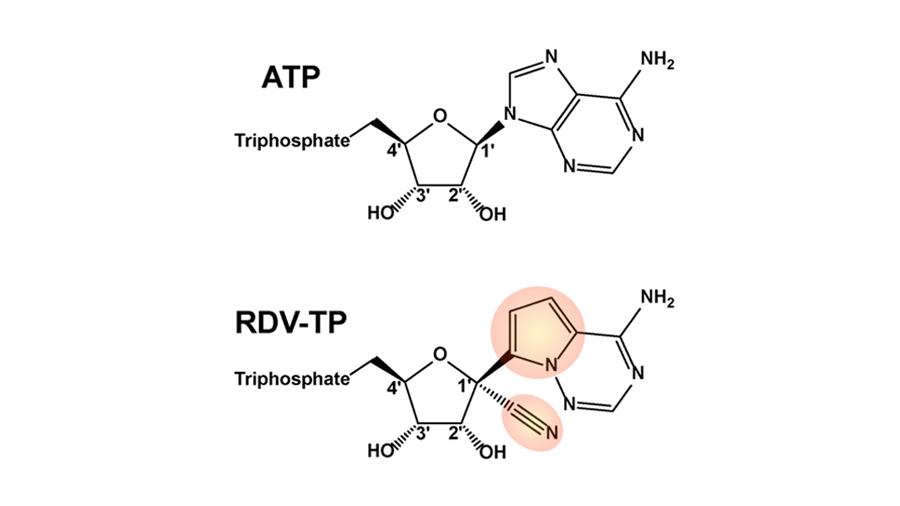
Evading viral proofreading
This is where remdesivir comes in.
“From my perspective, the (remdesivir) story started in 2013,” Denison said. “We had discovered that coronaviruses encode the only known RNA proofreading system … (and) I wanted to test whether there were any nucleosides out there that could be active in the setting of coronavirus proofreading.”
Denison heard from Cameron about a research collaboration with Gilead Sciences. Cameron was working on understanding the precise mechanism of action of a group of nucleotide analogs the company was using to treat hepatitis C, an RNA virus that infected tens of thousands of people each year.
Before the race to drug hepatitis C, according to Adrian Ray, a medicinal chemist who used to work at Gilead, “The nuc space for RNA polymerases and for RNA viruses was really not heavily explored.”
In the course of trying to beat competitors to the lucrative hepatitis C market, Gilead had developed a large library of RNA-dependent RNA polymerase inhibitors, including the molecule that would come to be known as remdesivir. A compound closely related to remdesivir had made it to early clinical tests for hepatitis C, but had faltered, in part because like remdesivir it needed to be administered by injection. Gilead changed strategies, buying a biotech startup to gain access to the startup’s orally available nucleotide analog, which became a key component of Gilead’s hepatitis C cocktail.
Working with the RNA-dependent RNA polymerase from poliovirus, Cameron had begun work that would show that the molecule was a non-obligate chain terminator, a type of nucleotide analog that the polymerase ought to be able to incorporate into a growing strand and keep going — but could not.
Intrigued, Denison contacted Gilead to ask permission to try that approved drug, called sofosbuvir, against mouse coronaviruses.
“That was their world-changing drug that cured hepatitis C,” Denison said. “They weren’t going to let a little-known virologist working on a coronavirus use their drugs.” But after a series of introductions by Cameron and several discussions, Gilead agreed to let his lab work with a different series of molecules, the ones that had been developed in-house and shelved for hepatitis C. They had shown promising results in early studies as a candidate treatment for other viral infections, including against Ebola virus.
The candidate molecules arrived. Denison and his trainees had no idea what they were. But they went ahead and tested them. What they found was exciting: In mouse cell culture, the drugs could block coronavirus replication.
“So we asked our Gilead collaborators, ‘What is that (compound)?’ They said, ‘We’re not going to tell you, but we’re going to send you 60 prodrugs, chemical modifications of the same compound,” Denison said.
One of that second batch of molecules proved to be remdesivir. Graduate student Maria Agostini, who had recently joined Denison’s lab, was one of the researchers who worked on understanding its strong activity.

“We’ve worked with a couple of potent compounds, but remdesivir was really one of the first I had worked with,” Agostini said. “When you were looking at the cells, you could visually see less evidence of viral replication going on.”
Whereas cells in her control dishes were visibly infected, suffering bad cytopathic effects, the cells treated with remdesivir after infection survived well. By growing many generations of the virus in cells treated with subtherapeutic concentrations of remdesivir, selecting for mutations that would let the virus evade the drug, Agostini and colleagues Erica Andres and Clint Smith demonstrated that it would take mutations in the viral polymerase to confer resistance to remdesivir — and that those mutant viruses were less able to infect hosts than the wild type.
Gilead supplied the drug free of charge, and the National Institutes of Health funded the researchers through a program aimed at developing treatments for emerging infectious diseases.< /p>
“This points out the value of collaborative science,” Denison said. “This was a company that committed to helping us do this when no one was interested in coronaviruses, and a grant mechanism that allows some flexibility in terms of expanding it.”
Recently, Mathias Götte’s enzymology lab in Canada has looked at how remdesivir works on polymerase enzymes from the coronavirus that causes MERS. Researchers in Götte’s group determined that the compound stops the polymerase, acting as a chain terminator — but not immediately.

Andrea Pruijssers, a virologist who directs the antivirals research program in Denison’s group, said, “(Remdesivir) somehow evades recognition by the proofreading enzyme.”
Instead of stopping the polymerase as soon as it is incorporated, remdesivir seems to let the enzyme keep going for a few more cycles but then causes it to stall. Researchers suspect that the molecular stumble may be caused by an unusual structure in the template-copy RNA duplex.
“They think that, at that point, the nucleoside analog that has already been incorporated is shielded from the proofreading enzyme,” Pruijssers said.
In the lab of Denison’s longtime collaborator Ralph Baric at the University of North Carolina, Chapel Hill, researchers found that remdesivir was an effective treatment for mice infected with SARS. Those results were promising enough to advance remdesivir into a study of MERS in monkeys, published in February by researchers at the Rocky Mountain Laboratory of the NIH. The drug showed some benefit in reducing the severity of the illness, provided the monkeys were treated prophylactically.
In terms of drug development, remdesivir “has sort of met every milestone along the way, from our perspective,” Denison said.

Similar drug class, different result
When Pruijssers started in the Denison lab in 2017, they had another molecule to investigate, beta-D-N4-hydroxycytidine, or NHC for short, that was being tested at the Emory Institute for Drug Development as a potential broad-spectrum antiviral drug.

Agostini led the first study of that drug as well, showing its activity in tissue culture. In March, researchers from the Baric and Denison labs published a followup in Science Translational Medicine, showing that NHC can block replication in the viruses that cause MERS and SARS — and also the coronavirus that causes COVID-19.
“It’s interesting,” Pruijssers said. “(NHC) doesn’t act as a chain-terminator. It incorporates into the genome and then causes lethal mutagenesis.”
Through its ability to base-pair with more than one nucleotide in the complementary strand, NHC introduces a cascade of errors in successive rounds of replication. Eventually, viral progeny don’t have the information they need to make a new virus.
Whereas remdesivir stops the polymerase in its tracks, causing few new mutations, deep sequencing experiments in the few viruses that emerged after NHC treatment showed that the drug causes numerous mutations.
“We identified two compounds that structurally fall into the same class of nucleoside analogs but act very differently on coronaviruses,” Agostini said.
They were particularly encouraged because the work showed that NHC has some therapeutic efficacy in the mouse model of MERS — and that it can block even remdesivir-resistant strains of coronavirus.
In an interview in early March, Pruijssers said NHC was still relatively untested as a therapeutic. “It’s hard to develop a mutagen, because the FDA doesn’t usually like the idea of mutation unless it’s for a life-threatening disease.”
But the pandemic changed things. Emory has partnered with Miami biotechnology company Ridgeback Biotherapeutics to develop the molecule, and Emory has filed an application with the U.S. Food and Drug Administration for permission to begin first-in-human trials to test the molecule’s safety.
Denison described the pivotal role Agostini played: “In five years of graduate school, Maria tested and developed the detailed in vitro analysis on two potential drugs to treat this pandemic coronavirus — and got them all the way through that in vitro preclinical development.”
It’s a remarkable and highly unusual feat. Drug development is known for its high failure rate. Of course, many drug candidates that appear promising in preclinical studies falter in large human trials.
Facing the pandemic
After arguing for a decade that the world must be ready for a pandemic, Denison said in early March, it was strange to be facing it.

“It’s really weird that we worked on this for the past six years, and the drugs were just getting through, and they were just ready to go,” he said. “We’ll see what the outcome is.”
He has some concerns about how to interpret data from clinical trials testing how well remdesivir works for COVID-19 in humans, the first of which are expected to be released this month as a trial at the China–Japan Friendship Hospital in Wuhan concludes. (Other trials, sponsored by Gilead, the NIH and the World Health Organization, launched later.)
First, animal data from other coronaviruses suggest that the drug is most effective when it’s delivered prophylactically, after the animals are exposed but before they begin to develop symptoms. In the context of a global pandemic, this would be difficult to achieve in humans. At some point in the course of infection, an extremely strong immune response begins to do more harm than the virus; at that point, Denison said, it may be too late for an antiviral to help.
Second, the data from the first few trials are likely to be nuanced and require careful interpretation — which Denison worries public discourse is not well prepared for. Early human trials of remdesivir’s sister compound in hepatitis patients showed dramatic differences among individuals in antiviral response, and remdesivir itself showed limited benefit compared to other candidate therapies in a clinical trial in Ebola patients. “People tend to have a winner or loser mentality,” he said. “They’ve called remdesivir ‘that failed Ebola drug,’ right? It didn’t fail in the Ebola trial; it just wasn’t advanced because it didn’t show as much benefit as the other two compounds.”
Whatever the outcome of clinical trials of remdesivir and NHC, Denison said he hopes this crisis will underline the importance of funding research into potentially pandemic viruses before outbreaks begin.
“Trying to maintain basic investigations and drug development against something that’s a high, high, high impact but low, low, low probability is really hard in our world. Really hard,” he said. “We’ve just never given up on this idea that we had to have these things ready and have them in the bucket.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.

Building a better model for drug delivery across the blood–brain barrier
Industry and academic scientists collaborated to develop a rat with humanized iron-transport receptors, enabling research into iron homeostasis and drugs that cross the brain’s barrier.

Fat synthesis enzyme crucial for milk fat and newborn growth
Researchers found that a deficiency of the fatty acid synthesis enzyme stearoyl-CoA desaturase-1 reduced mammary gland function during lactation and caused low birth weight in newborns that were fed milk from enzyme-deficient glands.