JBC: Team effort to figure out a rare genetic disease

About three years ago, Kristin Lindstrom was stumped. The medical geneticist, then based at the University of Rochester, had a patient referred to her by an endocrinologist. The endocrinologist was wondering if the patient’s unusual combination of features could be attributed to an underlying genetic syndrome.
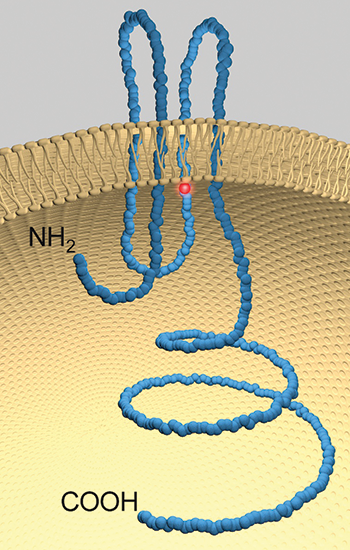 A model of pannexin 1 with the location of the mutation highlighted in red.DALE LAIRD
A model of pannexin 1 with the location of the mutation highlighted in red.DALE LAIRD
But the 17-year-old patient had features that didn’t fit any descriptions of known genetic disorders. The teenager was intellectually disabled, unable to read or count past 30, and needed help with daily activities. Her ovaries failed to develop normally. She had severe hearing loss that required cochlear implants. Her back was growing hunched.
When Lindstrom’s team did an evaluation of the young woman’s chromosomes, they didn’t see anything out of the ordinary. “We started thinking about the possibility of a single-gene disorder. I searched genetic reference books and online databases, but she really didn’t fit any one syndrome perfectly,” recalls Lindstrom.
So, in 2014, a year after Lindstrom first met the patient, her team decided to have the DNA taken from the young woman and her parents analyzed by whole-exome sequencing. Whole-exome sequencing searches for the parts of the genome that make proteins. Any variations in those protein-coding sequences could be mutations that cause a disease.
People have two copies of every gene, one inherited from each parent. The results from whole-exome sequencing showed a mutation in a particular gene. The woman’s parents each had one normal copy and one copy with the mutation. Both copies of the patient’s gene had the mutation, indicating the inheritance of a disorder that only manifests when both copies are defective. The mutation was in a gene that made a protein called pannexin 1.
But what was pannexin 1? “Often the results of exome testing are for conditions that we already know something about,” says Lindstrom, who since has moved to the Phoenix Children’s Hospital. “But this was different.” The young woman appeared to be the first patient to be reported with the mutation in the gene for pannexin 1.
Lindstrom had to turn to PubMed, the database of scientific papers. She began to hunt for scientists who would know about this protein and be willing to work with her team to find out more about the mutation.
That is how Dale Laird, a cell biologist at the University of Western Ontario in Canada, came to receive an email from Lindstrom about two years ago. Laird’s research interests were on gap junction proteins. These proteins let neighboring cells exchange information, in the form of molecules, with one another. In 2000, a new family of channel proteins was discovered that initially seemed to be like gap junction proteins. Called pannexins, these proteins were later shown to be channels that release ATP and other small molecules.
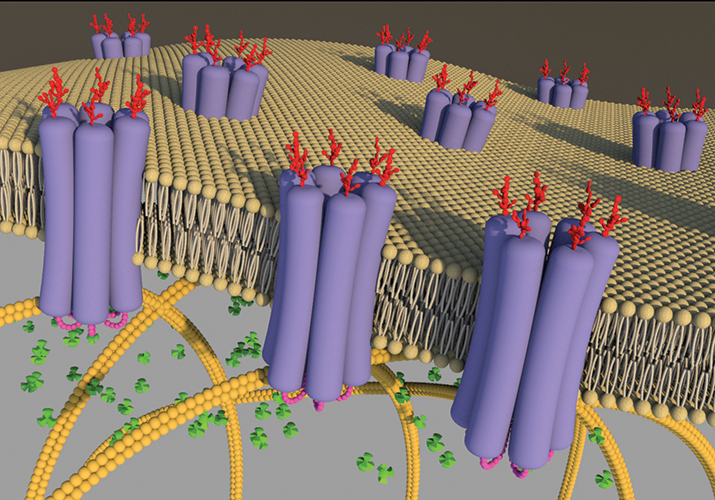 Schematic image of Panx1 channels in the lipid bilayer. It shows oligomers of six glycosylated Panx1 subunits arranged into large-pore channels that are suitable for allowing the passage of ATP (green cloverleafs). Permission to reproduce image was given to Laird and reproduced from an article by Penuela. S. et al., Biochem J 461,371-381 (2014).
Schematic image of Panx1 channels in the lipid bilayer. It shows oligomers of six glycosylated Panx1 subunits arranged into large-pore channels that are suitable for allowing the passage of ATP (green cloverleafs). Permission to reproduce image was given to Laird and reproduced from an article by Penuela. S. et al., Biochem J 461,371-381 (2014).
More intriguingly, pannexins appeared to be in almost every tissue of the body. Because of their ubiquity, pannexins were implicated in diseases as diverse as Crohn’s, osteoarthritis and cancer. “They’ve even been proposed to be hijacked by the HIV infection processes,” says Laird. Laird’s group became interested in pannexins, focusing in particular on the biochemical properties of pannexin 1.
After exchanging emails, Laird and Lindstrom decided to collaborate to parse out how the mutation in the pannexin 1 gene found in the teenaged patient affected the protein. The investigators found the mutation in the gene corresponded to a change from an arginine to a histidine in a loop proposed to be tucked within the channel.
By analyzing the mutant channel in cell culture, the investigators found that the protein was properly made and sent to its usual place of residence, the cell’s plasma membrane. However, once it got to its destination, the channel was defective.
To characterize fully the improper functioning of the mutated protein, Laird and Lindstrom recruited the team of Michael Jackson at the University of Manitoba in Canada. Jackson’s group are experts in measuring the function of channel proteins. They showed that the mutant pannexin didn’t fully open, which explained why it was defective in releasing ATP.
The collaborators published their findings in the Journal of Biological Chemistry; their paper was selected as a Paper of the Week. But Laird says the work doesn’t fully answer the questions: Does the defect in pannexin 1 actually cause all the features in the 17-year-old patient? Are there other factors at play?
The only way to answer these questions, says Laird, is to find more patients with the same or other pannexin 1 mutations. With more patients, researchers can understand better which symptoms and findings come with having mutations in both copies of the gene that codes for pannexin 1.
Here, Lindstrom has taken the charge to contact other laboratories that do genetic testing through whole-exome sequencing to see if there are other patients with two mutations in this gene who have the same features as her patient. “We can’t really do much more clinically until the labs start identifying these patients. Nobody is sequencing people for this gene specifically yet, because, until our paper came out, most geneticists would have never heard about it!” she says. “You can’t test for what you don’t know exists.”
She points out that whole-exome sequencing is a relatively new technique so “only a small fraction of people have had their exome sequenced.” Lindstrom adds that she hopes that as the technology becomes more widely adopted, “we will have more people with similar mutations identified, and then it will be really interesting to see what these people all have in common.”
On his end, Laird’s next step is to dig deeper into the molecular and cellular effects of the mutated pannexin 1. He and his team are hoping to create a laboratory mouse that carries the mutated pannexin 1. He says, “The idea would be to see if a mutant mouse demonstrates some of the phenotypes that we see in the clinical presentations” in the teenage patient.
But Laird emphasizes the work is all about collaboration. “This is a team approach,” says Laird, noting that, for the JBC paper, every team brought in different skill sets that helped the full biochemical characterization of the mutated pannexin 1.
But he saves the biggest shout-out for Lindstrom: “If she didn’t contact me in the first place, we wouldn’t have gotten this off the ground.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Glaucoma model links immune signaling to disease progression
Researchers at Duke University determine genetic variations that could increase the risk of developing glaucoma.

Uncovering the molecular roots of fatty liver disease
Physician–scientist Silvia Sookoian discusses her path from hepatitis C care to MASLD research, her use of multi-omics to study steatotic liver disease, and how lipid metabolism and genetics are reshaping understanding of MASH and liver health.

Mitochondria shape kidney cell function
Researchers at the University of Washington, Seattle present the first quantitative comparison of mitochondrial interactomes between two epithelial cell types in the kidney.

Long-chain polyunsaturated fatty acids linked to postoperative delirium risk
Researchers show that altered lipid metabolism may contribute to postoperative delirium, a condition linked to increased risk for long-term cognitive decline. The study explores potential disease mechanisms, which have yet to be understood.

Glycosylation patterns across antibody isotypes distinguish tuberculosis states
Researchers at Taipei Medical University present the first site-specific glycosylation analysis of immunoglobulins in elderly tuberculosis patients.

Blood glycome possibly predicts lifespan
Researchers at the University of Santiago de Compostela show that total serum N-glycome can predict mortality independent of traditional risk factors.