Cystic fibrosis: current understanding and prospects

In 1939, pathologist Dorothy Anderson at the Babies’ and Children’s Hospital at Columbia Presbyterian Medical Center in New York examined autopsy findings of 49 children who had died from malnutrition and had been diagnosed with celiac disease.
Anderson described her findings as “cystic fibrosis of the pancreas,” thus identifying cystic fibrosis, or CF, for the very first time.

After her discovery, doctors and researchers noted the genetic incidence of CF in families, prompting the discovery of a Mendelian autosomal recessive mode of inheritance for the disorder.
Even now, 80 years later, there is no cure for the disease. However, thanks to research, treatments and initiatives by organizations such as the Cystic Fibrosis Foundation, those with CF can live long and productive lives.
May is National Cystic Fibrosis Awareness Month, which was started by the foundation. Each year, people come together to share their experiences and educate others about the disease and the realities of living with it. The theme this year is “Unity in Community.”
Neil A. Bradbury is a professor at Chicago Medical School at Rosalind Franklin University of Medicine and Science. He’s been working on understanding CF for more than three decades. (Full disclosure: He was my PI when I was in graduate school. See my author’s note below.)
I talked to Bradbury about what we know about the disease and its treatment so far.
What is cystic fibrosis?
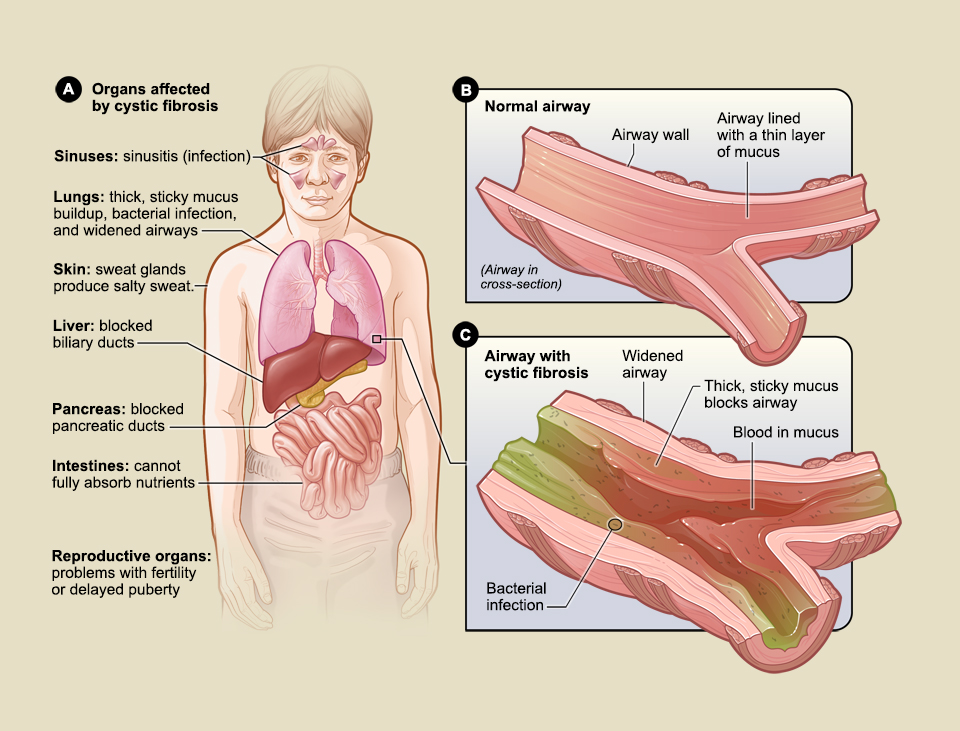
Cystic fibrosis is a genetic disease that occurs as a result of mutations in a gene which encodes a protein called cystic fibrosis transmembrane conductance regulator, or CFTR. It is a chloride and bicarbonate ion channel that helps to maintain salt and water balance in various tissues in the body.

The disease primarily affects epithelial tissues such as those lining the airways and the digestive and reproductive tracts. Specifically, it affects mucus-producing cells, meaning there is excessive mucus accumulation in tissues in CF.
In the lungs, this buildup of mucus clogs the airway cells, making breathing difficult; in the digestive tract, the ability of the pancreas to secrete digestive enzymes into the intestinal tract gets compromised. Thus, even though individuals with CF eat normal food, they are unable to access the nutrients.
“Generally, people with CF have problems with breathing and proper digestion,” Bradbury said, adding that even though most of the CF-affected population is of European decent, mutations in CFTR gene have been detected in people across the globe.
Early detection of CF
Because no cure exists for CF, it is important to detect the disease early and then to provide care and support. A breakthrough in CF disease management was the cloning of the cftr gene in 1989, which paved the way for early detection on CF in newborns. As a result, Bradbury said, “Certainly, in the U.S., every new-born undergoes a DNA test for CF, so most CF cases are picked up on neonatal screening.”
However, the DNA test is limited to the most common mutations in the gene, thus newborns harbouring selective familial mutations may escape the screening. To combat this, their blood samples are tested for a protein called immunoreactive trypsinogen, or IRT. This is a pancreatic enzyme precursor, blood levels of which are elevated in CF because of inability of the pancreas to release this enzyme into the intestine.
Bradbury’s journey toward understanding CF
As a graduate student at the University of Wales, Bradbury began to study salivary gland tissues of CF and non-CF individuals to understand the underlying mechanisms that lead to increased mucus secretion in CF patients.
“This was before the cftr gene was cloned, so we had no idea what gene or protein was involved,” he recalled.
Interestingly, his team found that mucus secretion from individual mucus-producing cells in CF was less, but salivary glands of CF patients had hyperplasia, meaning the number of cells producing mucus was more, thus leading to overall increased mucus secretion in CF phenotype.
Bradbury continued his research on CF through his postdoctoral stint and then as a principal investigator to understand the disease at a molecular level.
Soon after the cftr gene was cloned and it was discovered that the ion channel CFTR was responsible for CF, Bradbury’s team had what he called a “wild notion” that maybe CFTR undergoes a process called recycling, which means it moves in and out of the plasma membrane upon stimulation. At the time, no other ion channel had been shown to have this characteristic.
In 1992, his team discovered that CFTR in fact recycled quite frequently in response to stimulus. When their breakthrough research was published in Science, “it was very controversial and a lot of people dismissed it, but now it turns out that many other ion channels undergo the recycling process,” he said.
CF research: key focus areas
Considerable research has been done to understand the function of CFTR and how mutations in the cftr gene affect the protein function. This understanding has led to development of drugs that can treat CF.
Strategies to reverse CFTR defects is an active area of research.
“But,” Bradbury said, “there are also other things observed in CF patients for many years that cannot be explained by CFTR dysfunction.”
He points, for example, to alterations in lipid metabolism and microtubule dynamics. Bradbury said he it would be interesting to investigate what other proteins CFTR interacts with and how CF-causing mutations alter these interactions. This would help provide a holistic understanding of the disease pathogenesis.
CF therapy: standard of care and unmet needs
Mutations in the cftr gene can be broadly put into two categories: those that trap CFTR in the endoplasmic reticulum and prevent it from reaching the plasma membrane, which is its site of action, and those that allows CFTR to reach the plasma membrane but render the protein non-functional.
Accordingly, there are two kinds of drugs to treat CF: correctors that allow mutant CFTR to leave the endoplasmic reticulum and traffic to the cell surface and potentiators that improve the function of CFTR at the plasma membrane.
“Currently, the best medication for CF individuals is giving them a combination of correctors and potentiators,” Bradbury said, adding that this strategy has had a tremendous improvement in the lives of CF patients, who are “putting on weight, breathing better and living longer.”
However, even though the primary symptoms are CF patients are treated, individuals still require mucolytics to break down the mucus so that they can expectorate. Moreover, mucus attracts bacteria and other microorganisms, so CF patients undergo hospitalization from time to time for antibiotic therapy as many of them develop lung infection.
Additionally, longer life spans introduce a new complication: CF-related diabetes. CF patients have defects in release of pancreatic enzymes to metabolize glucose, as a result of which they develop this condition over time. “I am sure most patients would be quite happy to deal with diabetes rather than the more serious symptoms,” Bradbury quipped.
Looking ahead with certainty
Correctors and potentiators have been effective in CF treatment, and continuous improvements are being made on those drugs for the greater benefit. However, 5% of patients are unaffected by these drugs. These are cases where premature stop codons or alternative splicing result in truncated forms of CFTR, which are nonresponsive to approved medications.
Bradbury said scientists need to identify strategies to drive generation of full-length proteins to treat patients who are unresponsive to current therapy.
As means of cure, CRISPR-like technologies are also being explored to repair the defective cftr gene. This may gain more traction in the coming years with respect to improving vector delivery to cells of interest.
Bradbury said he also thinks that more physiologically relevant in-vitro models such as organoids would enable understanding of the molecular complexities of CF from an in-vivo perspective and help design better treatment strategies.
He noted that the Cystic Fibrosis Foundation has been “instrumental in setting up strong collaborations between research and clinical interventions for the benefit of patients, ”which is important for an effective bench-to-bedside transition for therapy. It is invested in ensuring that any basic research is translated into bedside treatment for patients. In fact, it’s approach is “seen as a model for how disease-based research foundations can improve the lives of patients,” Bradbury said. This had been adopted by other research foundations as well.
In the end, Bradbury said that he is hopeful that CF patients’ lives will only continue to improve in coming years.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Opinions
Opinions highlights or most popular articles

Learning can be fun: Gaming anatomy and physiology
Instructors explore how gamification and active learning transform student engagement and retention. They convey how emotion, interaction and design can make even rigorous subjects more effective and memorable.

Mentorship and uncertainty: Lessons from Telemachus
A biochemistry educator reflects on mentorship through the Greek story of Telemachus, showing how embracing uncertainty, failure and curiosity can transform teaching.

Embracing the twists and turns along the educator pathway
A biochemistry educator reflects on the challenges of early faculty life, describing how evidence-based teaching, cross-disciplinary collaboration and classroom challenges shaped her growth.

Redesigning with students in mind
Assistant professor reflects on how the shift to online teaching revealed gaps in points-based grading and led to a redesign centered on transparency and student growth.

Teaching beyond information transfer
Educator reflects on moving beyond lectures to create a biochemistry classroom centered on engagement, transparency and student ownership, showing how small shifts like “student hours” and active learning can transform understanding.

Mayday! Lessons from cellular dysfunction and group work dynamics
An upper-level biology course revealed that strong science doesn’t guarantee strong teamwork. One instructor shares how failed group dynamics reshaped their approach, leading to more structured, collaborative and effective student learning.