Crazy for cryo-EM

Cryo-electron microscopy is coming to a corner relatively near you. The National Institutes of Health has decided to fund national facilities with the latest cryo-EM instrumentation.
Although NIH-funded facilities won’t be springing up like Starbucks, the NIH, through a Common Fund initiative, is hoping to start off with three national service centers and eventually expand to having more around the U.S. The initiative aims to give researchers more opportunities to do molecular structure determination by high-resolution cryo-EM.

Why cryo-EM? The method, which has been around for 30 or so years, has had such a dramatic makeover recently that the journal Nature Methods bestowed it with the title “Method of the Year” in 2015. The once-fringe method is now beating the usual darling of structural biology, X-ray crystallography, at its own game, thanks to advancements in technology.
“We think — and this has been supported by many researchers in structural biology — that cryo-EM most likely will overtake or equal crystallography in the coming decade,” says Jon Lorsch, the director of the National Institute of General Medical Sciences. “The time seems right for a major investment to allow researchers in the U.S. to access these important new technologies.”
The NIGMS is administering the funding opportunity announcement for the Common Fund. The earliest date for submitting an application to host a service center is May 30. The earliest start date for the centers is projected to be April 2018.
For the veterans of cryo-EM, the move by the NIH is long overdue. Their microscopes have been in constant use, and they can’t keep up with the demand from other structural biologists who want to get in on the action. “People desperately want to get onto these instruments,” says Bridget Carragher at the New York Structural Biology Center. “If you don’t have access to one and your competitor does, you’re blown out of the water.”
Freeze!
To understand cryo-EM, think of the Mannequin Challenge of 2016. The social media phenomenon, which even had the Dallas Cowboys and Michelle Obama participating, involved people remaining stock-still in the middle of an action shot while a camera rolled and the song “Black Beatles” played.
The idea is more or less the same for cryo-EM: Researchers flash-freeze their samples at liquid-nitrogen temperature so that the molecules remain frozen in action, and then researchers use high-end cameras to capture the images of the molecules. (The song is missing.) By looking at the frozen action shots of the molecules, researchers can discern their structures and functions.
The method of freezing molecules to study them by electron microscopy made its way into biological applications in the 1980s. In the early 1980s, Jacques Dubochet, then at the European Molecular Biology Laboratory and later at the University of Lausanne in Switzerland, described the plunge-freezing method of sample preparation (the “cryo” part of “cryo-EM”). The rapid freeze, going from room temperature to −195.79 °C (the temperature of liquid nitrogen) in fractions of a millisecond, causes water to form vitreous ice, which is an amorphous solid form of water that doesn’t have ice crystals. The paper is noted as the work that helped microscopists embrace cryo-EM.
The thin layer of vitreous ice holds molecules more or less in the way they would be held in aqueous solution, so the freezing process allows researchers to see molecules practically in their native state. Cryo-EM doesn’t require the protein to be forced into a crystal, which is a bottleneck in X-ray crystallography. The method also doesn’t require as much purified sample as does X-ray crystallography. Richard Henderson at the Medical Research Council points out that the protein sample “doesn’t need to be particularly pure … 90 percent pure would be perfectly fine.” The method also can handle proteins as small as 100 kilodaltons and ones as large as viruses.
Large molecules contain more signals and so are easier to detect than small molecules, says Erhu Cao at the University of Utah, adding that “there is no upper limit” on what can be viewed by cryo-EM. That’s why large complexes, such as ribosomes and viruses, have been among the first structures to be solved by cryo-EM. For example, the structure of the Zika virus was solved in 2016 by two independent groups using cryo-EM.
Most importantly, cryo-EM can visualize protein complexes and give indications about protein flexibility. As the freezing process captures the molecules in action, cryo-EM permits researchers to look at multiple conformations of molecules within a single field of view and get an appreciation for the dynamics of a protein. When looking at protein complexes, explains Jeff Lengyel at the company FEI, low-affinity interactions are easier to discern by cryo-EM. FEI, which was bought out by Thermo Fisher Scientific, is a vendor of cryo-EM instrumentation,
See clearly now
Instead of using light to image molecules, as is done in optical microscopes, researchers use beams of electrons in cryo-EM. The way the electrons bounce off a molecule gives a picture of the molecule. The scattering of electrons as they hit the frozen molecules is picked up by detectors.
The detectors are the reason for cryo-EM’s sudden surge in popularity. For most of cryo-EM’s existence, researchers used film. “We collected data on film that we would have to develop the old-fashioned way in darkrooms,” recalls Eva Nogales at the University of California, Berkeley. “That made it very difficult to automate the data collection and (achieve) high throughput.”
Charge-coupled device cameras came along next. “They were great for high throughput but they never got to high resolution,” notes Nogales. The CCD cameras detected electrons in a roundabout way. The cameras were coated with a film of phosphoscintillator, which picked up only photons, not electrons. The electron signal had to be converted into photons of light, detected and then made into an image. The roundabout way didn’t help to get very crisp and clear images.
The introduction of direct electron detectors in 2013 changed the game. Their name says it all: “You are literally counting electrons, and that’s really transformative,” says Sriram Subramaniam at the NIH. “It essentially eliminates the noise that was present before in CCD images.”
The elimination of noise means crisper, clearer pictures of molecules, to the point of being able to see amino-acid side chains at resolutions as high as two or three angstroms.
 and β-tubulin (blue) determined by cryo-EM") The structure α-tubulin (green) and β-tubulin (blue) determined by cryo-EM IMAGE COURTESY OF EVA NOGALES
The structure α-tubulin (green) and β-tubulin (blue) determined by cryo-EM IMAGE COURTESY OF EVA NOGALES
Taking over structural biology
In the past four years, with the introduction of the direct electron detectors as well as advances in imaging software and instrument automation, it seems as though every week brings with it a new structure of a molecule solved by cryo-EM. (See box on “Tackling sample preparation.”) Several experts point to the structure of a mammalian TRP channel, TRP V1, as the structure that made structural biologists sit up and take notice.
TRP V1 is the protein that allows us to feel the heat, in the form of both temperature and the molecule capsaicin, found in hot peppers. The membrane protein resisted X-ray crystallography attempts for years, says Cao, who was tasked with solving the structure when he was a postdoctoral fellow in David Julius’ laboratory at the University of California, San Francisco.
Feeling defeated by his lack of success in solving the structure of TRP V1 by X-ray crystallography, Cao decided to give cryo-EM a shot. He got together with another postdoctoral fellow, Maofu Liao, in the laboratory of Yifan Cheng at UCSF in 2009. They spent almost three years optimizing sample preparation and imaging conditions for cryo-EM.
At that time, when Liao and Cao were working on the TRP V1 channel, others in the Cheng laboratory, along with the group of David Agard, also at UCSF, and the company Gatan were developing direct electron detectors and the software to accompany them. In 2013, Liao and Cao tested their sample preparation with the new detector.
The collaborators got a structure of TRP V1 at 3.4 angstrom resolution, “breaking the side-chain resolution barrier for membrane proteins without crystallization,” as they noted in the abstract of their 2013 Nature paper.
Cryo-EM had achieved similar or higher resolutions on other samples, but in all those cases there were structures of the same or similar proteins already solved by X-ray crystallography. TRP V1 was the first cryo-EM-solved structure that X-ray crystallography had failed to deliver. The TRP V1 structure proved that cryo-EM was catching up to the capabilities of X-ray crystallography.
Given some of the advantages that cryo-EM has over X-ray crystallography, many structural biologists started to dive into their freezers and to dig around for samples that they had given up on solving by X-ray crystallography. Now structures of ribosomes, viruses, ion channels and membrane proteins have been solved by cryo-EM.
And it’s not just fundamental science that benefits. Clinically important proteins now can be tackled by cryo-EM. Subramaniam’s group recently solved the structure of p97, which is a cancer target. “It’s a relatively dynamic protein,” says Subramaniam. “It’s been almost impossible to get structures of it by X-ray crystallography simply because of this fact. But we showed that, by classifying the images into different states, we could tease out three simultaneously present conformations, which is basically what made it difficult to get to by crystallography.”
Indeed, the ability of cryo-EM to help researchers deduce dynamics is a strong draw. Elizabeth Stroupe’s group at Florida State University is working on sulfite reductase. It’s an 800-kDa complex that can come together several different ways. She says, “The fact you now can do really accurate data collection means that, with enough biochemistry to back up our structure, even if we can’t get to atomic resolution, we should be able to start to understand these heterogeneous modes of action for this enzyme in a way that we couldn’t have done with crystallography.”
A more recent advent in cryo-EM has been phase plates, which have experts giddy with excitement. These are new bits of hardware, explains Lengyel. Phase plates help researchers overcome problems with sample damage and get enough information from their images with low-dose electron beams. “While it’s still very new, several labs are beginning to generate extremely promising results,” says Lengyel, whose employer, Thermo Fischer Scientific, sells the technology. “That will be the next advance in the field.”
Indeed, a paper showed up in bioRxiv in November that described the determination of the structure of hemoglobin at 3.2 angstroms with the use of phase plates. At this resolution, the investigators were able to see amino-acid side-chain densities and prosthetic heme groups. Hemoglobin has a size of 64 kDa, which dramatically drops the lower limit of the size of molecules that can be determined by cryo-EM.
Experts also are expecting cryo-EM to tackle samples more complicated than purified proteins. The method is now moving into the realm of visualizing proteins in their native environments inside cells.
 Eva Nogales and Robert Louder review a structure by cryo-EM. IMAGE COURTESY OF EVA NOGALES
Eva Nogales and Robert Louder review a structure by cryo-EM. IMAGE COURTESY OF EVA NOGALES
Economies of scale
With all the advances in cryo-EM, it’s not surprising there now is a clamor for access to the instruments. Experts interviewed for this story all expressed enthusiasm for the NIH funding opportunity announcement for the national facilities for cryo-EM but, in the same breath, said the NIH is playing catch-up. Lorsch accepts the criticism openly.
“The U.S. has been falling behind countries in Europe and Asia in terms of having access for researchers to these cutting-edge cryo-EM technologies,” he says. “We were very concerned about that and felt that something needed to be done. Francis Collins, as he was made aware of these issues, was very supportive of doing some significant investment in this area through the Common Fund.”
The Howard Hughes Medical Institute and the Wellcome Trust already have funded cryo-EM facilities to give researchers access to the instrumentation. The NIH has funded several cryo-EM consortia, but those are composed of cryo-EM experts. The National Cancer Institute opened a cryo-EM facility under the leadership of Subramaniam. But more are needed.
“Universities are coming to the conclusion if they want to maintain their standing in structural biology, they need to acquire this technology,” says Lengyel. “It’s getting to the point where X-ray crystallographers are either not accepting jobs or threatening to leave if they don’t get access to cryo-EM because it’s become so essential for their research.”
The investment into a cryo-EM setup isn’t trivial. As Carragher says, “It can’t be taken on as if it’s a Sunday-afternoon affair.”
Just the cost of a high-end instrument that can do high-resolution imaging is about $6 million. To run one instrument for one year costs half a million dollars. Then there are the costs for all the software, computers, hard drives for storage, electricity, air conditioning (so the computers and instrumentation don’t overheat) and people.
Some research institutions haven’t bothered to wait for the NIH and have gone ahead and set up their own cryo-EM facilities. But, as Lorsch points out, “Even if the institutions have enough capital to purchase an instrument, most of them will realize that, once they do so, the cost of maintaining it really becomes a major burden.”
The reason for the funding opportunity announcement is to, as Lorsch says, “create economies of scale. The old model is one in which each institution tries to buy its own major piece of equipment for everything, but we could really find a way to get more for the taxpayer’s money by creating these nationally shared facilities and get access for more people for the same amount of money.”
The expectation is that each center will have four microscopes, two that are high-end and two that are dedicated to screening samples for viability. There will be computational resources and data storage.
The precedent for this shared facility comes from the national synchrotrons that X-ray crystallographers use. Most crystallographers will make their crystals at their home institutions but then book a day or two at a national synchrotron facility, such as the ones at the National Institutes of Standards and Technology and Lawrence Berkeley Laboratory, to use the radiation source to collect data from their crystals.
The goal of the shared facilities is to give “biologists from all walks from structural biology access to this equipment and, very importantly, to give them training to become expert cryo-EM users,” says Lorsch. The NIH has noted that there is a dearth of cryo-EM experts to help other structural biologists enter the field, so the national facilities will work to train people to become experts in doing cryo-EM.
Even the instrument manufacturers are taken aback by the craze over cryo-EM. When FEI developed the Titan Krios, a high-end instrument, Lengyel says the company expected to sell 35 total worldwide. They sold more than 100. “The last couple of years have been the biggest explosion in the field, given the advent of the new detectors,” says Lengyel. “We expect that to continue.”
Lengyel isn’t the only one optimistic about the future of cryo-EM. “Very soon, there’s going to be very few labs that just do crystallography,” says Nogales. “That’s my prediction.”
Tackling sample preparation
Several experts point out that sample preparation for cryo-EM remains largely unchanged since the 1980s. “It’s not a very good method, because you waste a lot of protein,” says Yifan Cheng at the University of California, San Francisco.
To analyze a protein, researchers pipette a drop of about 3 microliters of their purified protein solution onto a carbon grid, wick off much of the drop with blotting paper and then plunge-freeze the grid with the thin layer of sample into liquid ethane that has been cooled by liquid nitrogen. “Everything in cryo-electron microscopy has improved since 1980 or so when it started except for the specimen preparation,” says Carragher. “We make the darn grids more or less the same way.”
Most of the sample ends up on the blotting paper that gets thrown in the trash can. To prevent waste of precious samples, Carragher’s group has developed a device called the Spotiton. The device uses piezoelectric dispensing to drop a few nanoliters, rather than microliters, onto a grid. The grid itself is self-blotting and wicks the sample into a thin layer. The device is en route to commercialization.
“If you can use tiny amounts of samples, many things become feasible,” says Cheng. “One thing limiting us is that biochemically, we still need to purify our protein sample. Purification is a major hurdle.” If researchers can get away with a few nanoliters of sample, it will make cryo-EM even more attractive.
 and Hui Wei (right) in the Carragher lab.") The Spotiton robot operated by Venkata Dandey (left) and Hui Wei (right) in the Carragher lab. PHOTO COURTESY OF BRIDGET CARRAGHER
The Spotiton robot operated by Venkata Dandey (left) and Hui Wei (right) in the Carragher lab. PHOTO COURTESY OF BRIDGET CARRAGHERThe structure of β-Galactosidase by cryo-EM. Image, whichis also on the cover, is courtesy of Veronica Falconieri and Sriram Subramaniam.
Enjoy reading ASBMB Today?
Become a member to receive the print edition four times a year and the digital edition monthly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles

Catching tau in the act
Using a new proximity-labeling approach, researchers reveal how tangles of the brain-associated protein tau may disrupt RNA biology long before neurons die.
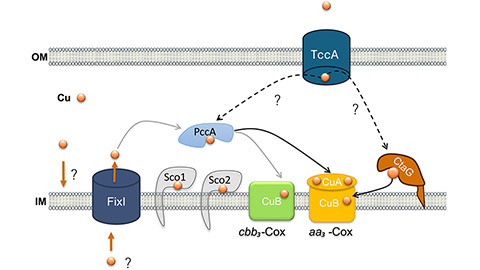
How copper delivery fuels bacterial respiration
Researchers identify the roles of several proteins in copper homeostasis in the aerobic bacterium Caulobacter vibrioides.
Revealing the glycoproteome of a cancer subtype
Researchers mapped the glycoproteome of extrahepatic cholangiocarcinoma and compared it to intrahepatic tumors. Differences in sugar modifications and immune cell content suggest new biomarkers and guide development of targeted immunotherapies.

Uncovering the mechanisms of a glycosylation disorder
Mutations in OGT, an enzyme that adds sugars to proteins, cause a rare neurological disorder. Using proteomics, researchers reveal how OGT interactions with TET proteins may trigger epigenetic changes and early neural defects.

Heat shock proteins as a promising breast cancer therapeutic
Researchers unveiled isoform-specific targets on heat shock protein 90 which may be beneficial in therapeutic development.

Optimized proteomic analysis of preserved biological tissue samples
Researchers have developed an optimized workflow for analyzing formalin-fixed paraffin-embedded tissue. This workflow provides an enhanced collection of unique proteins and phosphorylation sites for more detailed analysis of biological samples.