The drug of good and evil

Thalidomide is a molecule with a split personality. Notorious as the drug that caused thousands of babies to be born with grotesque deformities in the 1950s, it now has become the frontline defense for some cancer and immune-compromised patients. Until recently, researchers did not understand how this single molecule could have such extreme effects. But emerging research is showing that thalidomide acts at the molecular level in an unusual way, which could explain its Dr. Jekyll-and-Mr. Hyde effects.
Over the past four years, several groups have shown that thalidomide and drugs like it bind to a component in a ubiqutin ligase complex. Upon binding, thalidomide and its structural analogs change the proteins that the ubiquitin ligase targets for degradation.
The fact that thalidomide modifies interactions between proteins has been a revelation. Protein-protein interactions “have always been considered difficult to target with small molecules,” says Nicolas Thomä at the Friedrich Miescher Institute for Biomedical Research in Switzerland. When an established drug like thalidomide was shown to affect protein-protein interactions, Thomä adds, “that immediately caught our attention.”
As William Kaelin, a Howard Hughes Medical Institute investigator at the Dana-Farber Cancer Institute, notes, thalidomide has made researchers rethink how they can approach development of therapeutics. He says, “Thalidomide opens up a new paradigm for how you could drug proteins.”
Troubled history
Thalidomide first commercially appeared in 1957 when a German company called Chemie Grünenthal, later known as Grünenthal, began marketing the drug in Europe to treat morning sickness in pregnant women as well as insomnia. According to a 2013 New York Times story, the drug was so in demand in some European countries that it became nearly as popular as aspirin. Soon, 40 countries around the world, though the United States was not among them, had made thalidomide available.
But the ominous signs quickly appeared. The first known victim of thalidomide was a girl born on Christmas Day of 1956 to a Grünenthal employee. The baby had no ears. By 1960, more than 10,000 women who had taken the drug while pregnant had given birth to babies with ghastly physical deformities, including spinal cord defects and flipperlike arms and legs. About 40 percent of the babies, known as “thalidomide babies,” are thought to have died within the first year of birth.
Few of these babies were born in the U.S. In 1960, a new employee joined the U.S. Food and Drug Administration. As an ease-into-the-job type of project, new hire Frances Oldham Kelsey was asked to review the application for thalidomide for approval for use in the U.S. As Kelsey looked through the information supplied by Grünenthal and its U.S. distributor, William S. Merrell, she grew concerned about the lack of safety information for the drug. Kelsey doggedly pursued more safety data, which led to the outing of evidence in Germany in the fall of 1961 that linked birth defects to the drug. The next year, the drug was withdrawn from the global market.
The scale of the disaster wrought by thalidomide was so large that the World Health Organization launched its International Drug Monitoring program in 1968. Grünenthal apologized to thalidomide victims in 2012.
Discovering other uses
Although thalidomide had become the poster child for a drug gone wrong, doctors continued to prescribe it – but for different reasons. In 1964, an Israeli doctor at the Hadassah University Hospital, Jacob Sheskin, prescribed thalidomide for a leprosy patient’s insomnia. The patient had a painful reaction to leprosy called erythema nodosum leprosum, which brings on fever, weight loss and arthritis. Much to Sheskin’s surprise, the patient’s condition dramatically improved. Sheskin then tried the drug on several other patients and got the same result.
The WHO carried out a trial in the 1970s and determined that thalidomide was then the best treatment option for erythema nodosum leprosum. In 1998, Celgene won FDA approval to market the drug to treat skin lesions caused by leprosy. The drug was administered with stringent controls to make sure female patients did not become pregnant during the course of the treatment. However, once other drugs, such as prednisolone, were shown to be more effective for treating leprosy, the WHO no longer supported the use of thalidomide. Brazil is one country that continues to use thalidomide to treat leprosy. About 1,000 Brazilian babies have been born with thalidomide defects in the past two decades because of inadequate supervision during the drug’s administration.
In 2000, at a meeting of the American Society of Clinical Oncology, Bart Barlogie at the University of Arkansas for Medical Sciences reported that thalidomide slowed the progression of multiple myeloma in people who had failed to respond to conventional therapy. After sufficient case studies accumulated, the FDA extended its approval of thalidomide to treat multiple myeloma in 2006.
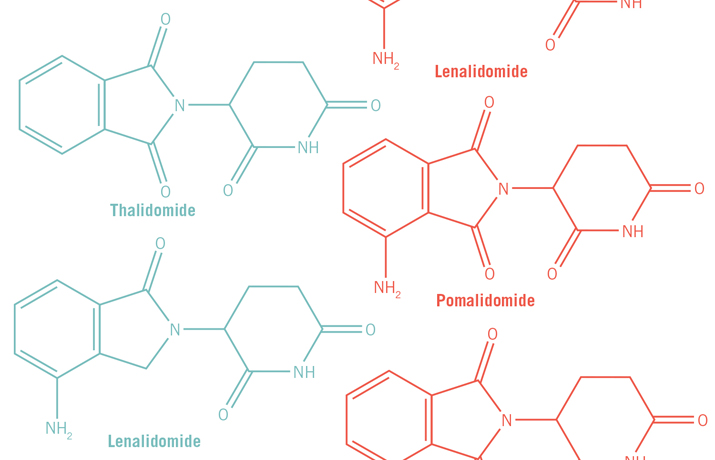
Currently, Celgene markets thalidomide along with two structural analogs it has developed, lenalidomide and pomalidomide. All three are approved by the FDA to treat multiple myeloma and have become frontline drugs for treating the relapsed and more resistant cases of the disease. Multiple myeloma is a cancer caused by malignant plasma cells. Under normal circumstances, B cells mature into plasma cells, which are immunoglobin factories. But when plasma cells become cancerous, they spin out of control and produce bone tumors. A patient has multiple myeloma when he or she has more than one tumor. Though structurally similar, lenalidomide and pomalidomide are more efficacious than thalidomide in treating multiple myeloma. The three drugs are known collectively as immune modulating drugs, or IMiDs.
Mysteries of the molecular mechanism
The mechanism by which thalidomide and the other two IMiDs acted was not understood until Hiroshi Handa at the Tokyo Medical University and colleagues published a paper in the journal Science in 2010. In that paper, they identified a protein called cereblon that was bound by thalidomide. Cereblon, which is ubiquitously expressed, belongs to an ubiquitin ligase complex. Thomä says there were many publications on a plausible mechanism for thalidomide’s mode of action, but Handa’s team was the first to pull out cereblon. “They, without a doubt, established thalidomide as a molecule that binds ubiquitin ligase,” he says.
The finding made researchers sit up for two reasons. Conventional thinking had dictated that drugs couldn’t target ubiquitin ligases, because the enzymes were much too essential for protein degradation and turnover. Also, scientists had assumed the ubiquitin ligase complex had too many protein-protein interactions within it for a drug to target one of the interactions specifically.
But here was thalidomide, developed some 50 years ago on solely phenotypic screens, capable of binding to a part of a ubiquitin ligase complex. The binding hinted that “some effects that you see of thalidomide, good and bad, have something to do with the ubiquitin ligase” modifying its function, says Thomä.
Other studies followed using the other two analogs of thalidomide. Two teams, one led by Kaelin and the other by Benjamin Ebert at Harvard Medical School, independently showed earlier this year that the ubiquitin ligase is able to target two zinc-finger transcription factors, IKZF1 and IKZF3, for degradation when lenalidomide is bound to cereblon. A group from Celgene also published the same result. The finding began to give clues as to why IMiDs work against multiple myeloma.
IKZF1 and IKZF3 have opposite effects on the survival of T and B cells of the immune system. In the case of T cells, IMiDs help the cells survive by downregulating IKZF1 and IKZF3. When those two transcription factors are expressed, they repress the expression of the interleukin-2 gene in T cells and keep the cells dormant. However, when IKZF1 and IKZF3 are degraded, which is what happens when an IMiD binds to the ubiquitin ligase, interleukin-2 gene expression triggers the stimulation of T cells.
In contrast, IMiDs cause malignant B cells to die. The presence of IKZF1 and IKZF3 normally promotes the development of the B cells into mature, antibody-producing plasma B cells. The degradation of IKZF1 and IKZF3, which, again, is what happens when an IMiD binds to the ubiquitin ligase, triggers apoptosis of B cells. In myeloid myeloma, B cells turn malignant, so that’s why IMiDs are effective in treating the disease.
“It’s always been a paradox,” says Rajesh Chopra of Celgene of the IMiDs’ ability to kill malignant B cells but trigger T-cell stimulation. But the emerging picture of the mechanisms of action of IMiDs on IKZF1 and IKZF3 “explains that duality of function.” The differences in effect, says Chopra, allow researchers to start thinking about how to exploit the variations in protein networks inside cells with a single drug.
Finding more puzzle pieces
This summer, Thomä and colleagues reported the crystal structure of thalidomide bound to cereblon. That work revealed another aspect of thalidomide’s mechanism of action: When the drug binds to cereblon, the protein can’t bind to one of its usual targets, the homeobox transcription factor MEIS2. Downregulation of MEIS2 in chicken embryos at the proximal limb-bud region has been shown to be important for limb development.
Putting together the puzzle pieces that they have so far, researchers think that IMiDs modify cereblon’s choice in binding partners, making it turn to new substrates like IKZF1 and IKZF3 at the cost of losing MEIS2 and possibly other native substrates. “Here’s an example where the drug bound to the protein alters – in a qualitative way – the function of the protein,” says Kaelin.
But researchers stress that they still don’t know which substrate causes thalidomide’s ugly side effects. “The story is not simple,” cautions Handa. “Maybe other substrates might be involved.”
Handa’s group, in collaboration with researchers from Celgene, also has produced a crystal structure of lenalidomide bound to cereblon and one of its binding partners in the ubiquitin ligase complex. The structure shows how three tryptophan residues in the C-terminal domain of cereblon are critical for binding to one part of the IMiD’s structure.
The structures collected by various research groups also showed that another part of the drug partially sticks out from the complex into solution, hinting that this exposed portion of the drug could be responsible for the altered preference of substrates targeted for ubiquitination.
“Since part of the molecule is exposed in the putative substrate-binding region, the small molecules themselves may form part of the interface with the substrate adaptor and the substrate,” says Ebert, who has consulting and research collaboration ties with Celgene. The molecules “may directly modulate what proteins bind to the cereblon and therefore what proteins get ubiquitinated and degraded.”
Thalidomide, lenalidomide and pomalidomide all bind to the same site on cereblon at its C-terminal region. But because they have a number of differences in the part of the structure that sticks out into the solvent, researchers say that differences at the atomic scale, at one or two carbon sites on the rings of the drugs, may explain the differences in efficacy of the three drugs for treating multiple myeloma. The finding that a few atomic changes make a significant impact on the efficacy of the different IMiDs “highlights that the iMIDs are not the same,” says Chopra. Indeed, Celgene scientists insist that lenalidomide and pomalidomide not be lumped together as “thalidomide analogs.” “They look structurally the same, but the substrate of consequence may be different,” says Chopra.
Hope for more
The recent findings are exciting, say the researchers, because they suggest that drugs developed down the road may be able to modify the activity of a protein complex rather than simply target it for disruption. “It is perhaps the most exciting aspect of the whole field now,” says Ebert. “It’s a completely novel mechanism of action for a drug, where the presence of a drug modulates the substrate specificity of the ubiquitin ligase.”
But fundamental questions remain.
For example, although thalidomide causes limb deformations in humans, zebrafish and chickens, it doesn’t cause fetal defects in mice or rats, Handa points out. This prompts the question: What is different about the rodent and murine forms of cereblon that do bind thalidomide but somehow bypass its devastating effects?
Researchers also point to the 1995 work of Robert D’Amato’s group at Boston’s Children Hospital, which demonstrated that the drug inhibited the growth of blood vessels and had antitumor activity. No one has yet figured out what causes the drug’s antiangiogenic effects or, for that matter, its sedative effects, which is the effect that put thalidomide on the world stage in the first place in the 1950s.
“There’s a bit of a discovery phase in place right now so that we better understand the range of functions that cereblon can assume when occupied by different IMiDs,” says Celgene’s Tom Daniel. And Ebert adds, “There is an enormous hope that this could be the beginning of a new class of drugs that induces degradation of a variety of disease-relevant proteins.”
Enjoy reading ASBMB Today?
Become a member to receive the print edition monthly and the digital edition weekly.
Learn moreGet the latest from ASBMB Today
Enter your email address, and we’ll send you a weekly email with recent articles, interviews and more.
Latest in Science
Science highlights or most popular articles
Cows offer clues to treat human infertility
Decoding the bovine reproductive cycle may help increase the success of human IVF treatments.
Immune cells can adapt to invading pathogens
A team of bioengineers studies how T cells decide whether to fight now or prepare for the next battle.
Hinton lab maps structure of mitochondria at different life stages
An international team determines the differences in the 3D morphology of mitochondria and cristae, their inner membrane folds, in brown adipose tissue.

National Academies propose initiative to sequence all RNA molecules
Unlocking the epitranscriptome could transform health, medicine, agriculture, energy and national security.
From the journals: JLR
What can you do with artificial lipoproteins? A new key to angiogenesis. Flavonoids counteract oxidative stress. Read about recent papers on these topics.
Iron could be key to treating a global parasitic disease
A study has found that leishmaniasis causes body-wide changes in iron balance, leading to red blood cell damage.